Department of Marine Ecology, Centre for Advanced Studies of Blanes (CEAB, CSIC), Blanes, Catalonia, Spain.
Department of Evolutionary Biology, Ecology and Environmental Sciences, and Institute of Biodiversity Research (IRBio), University of Barcelona, Barcelona, Catalonia, Spain.
Ecol Appl. 2020 Mar;30(2):e02036. doi: 10.1002/eap.2036. Epub 2019 Dec 11.
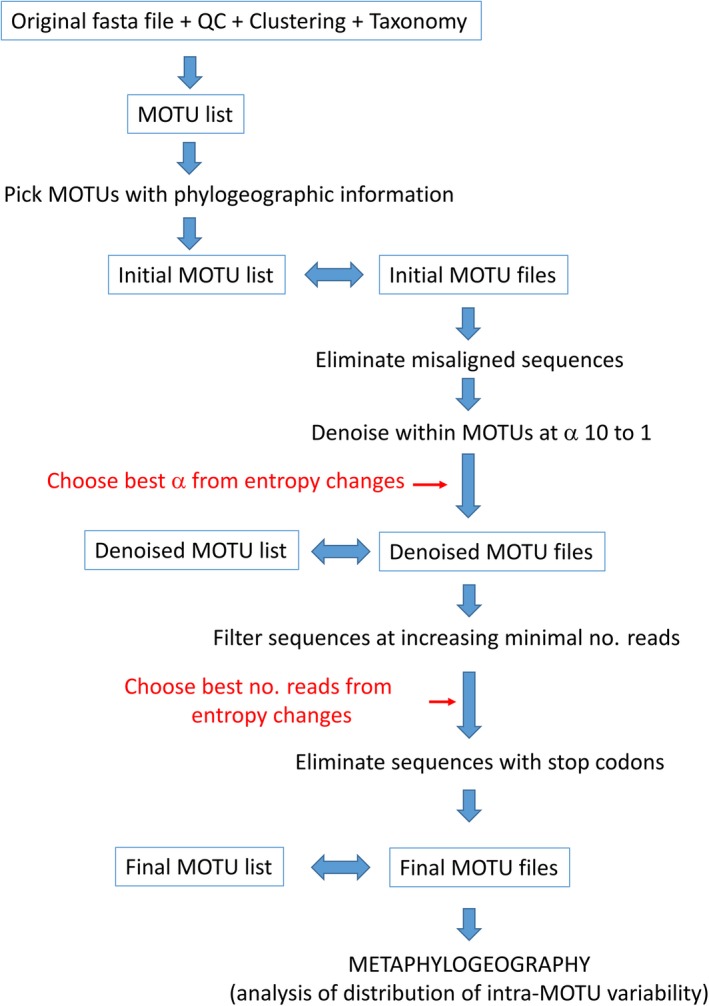
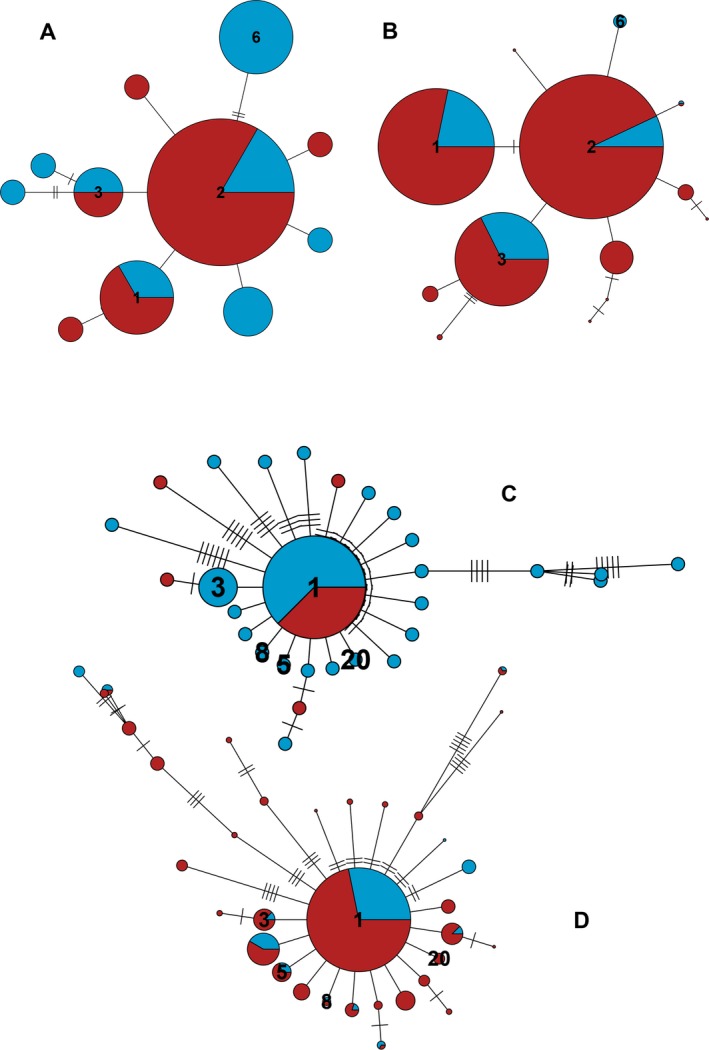
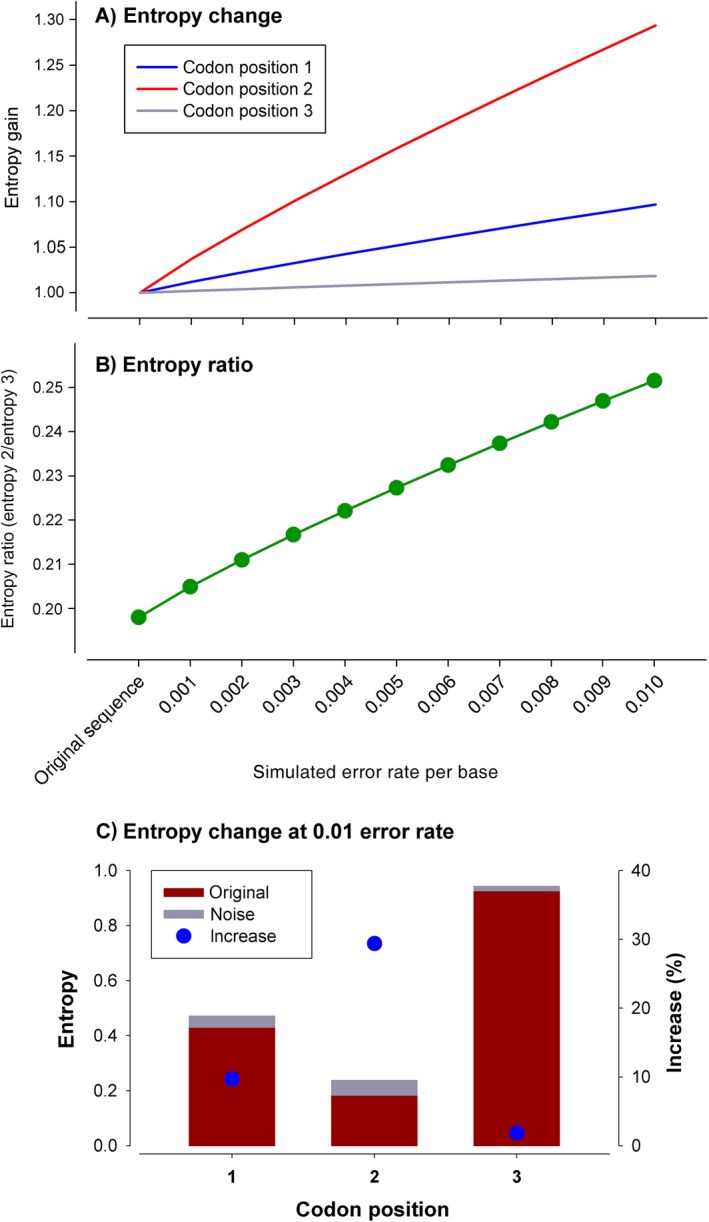
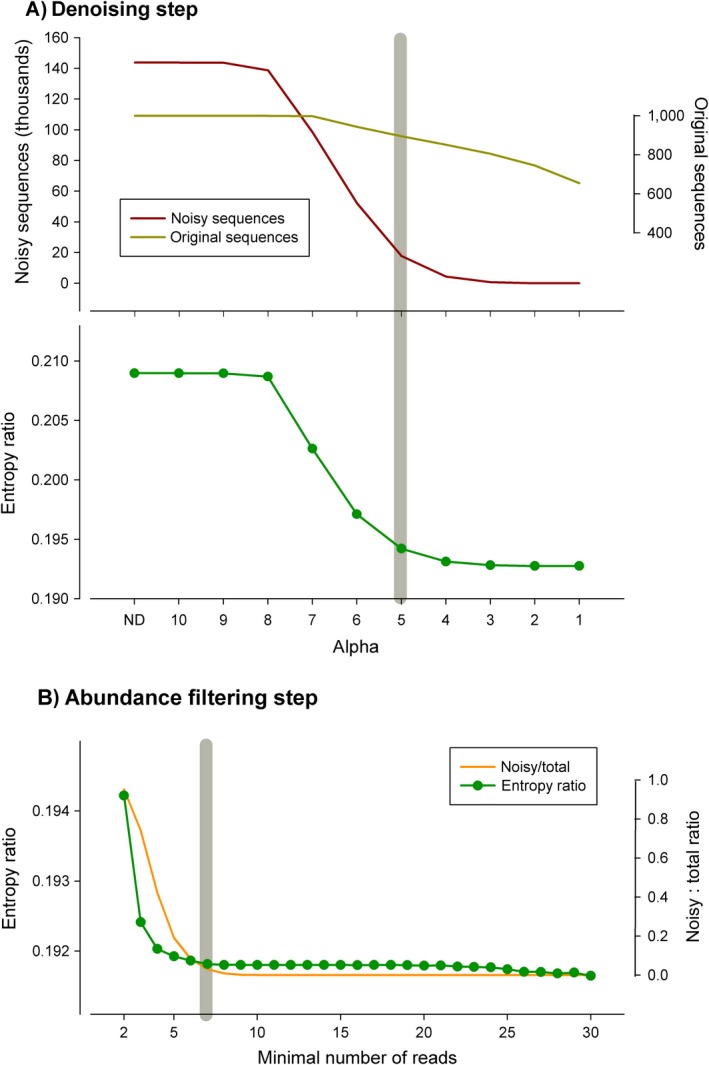
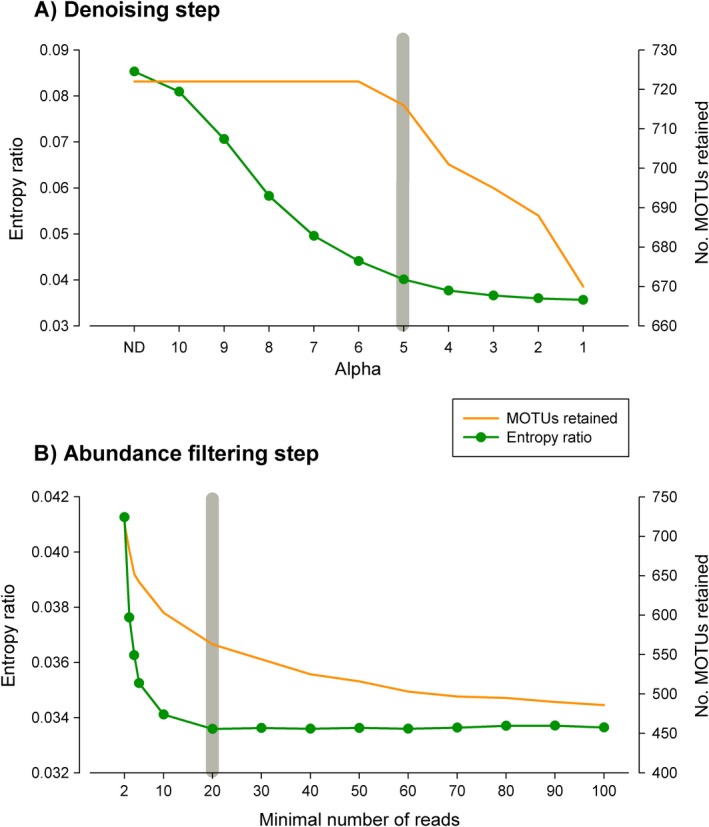
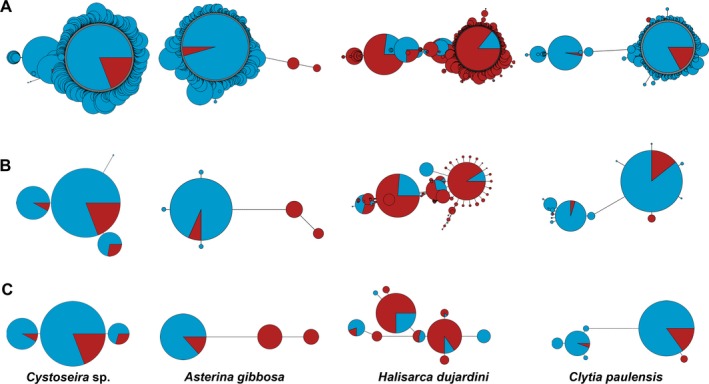
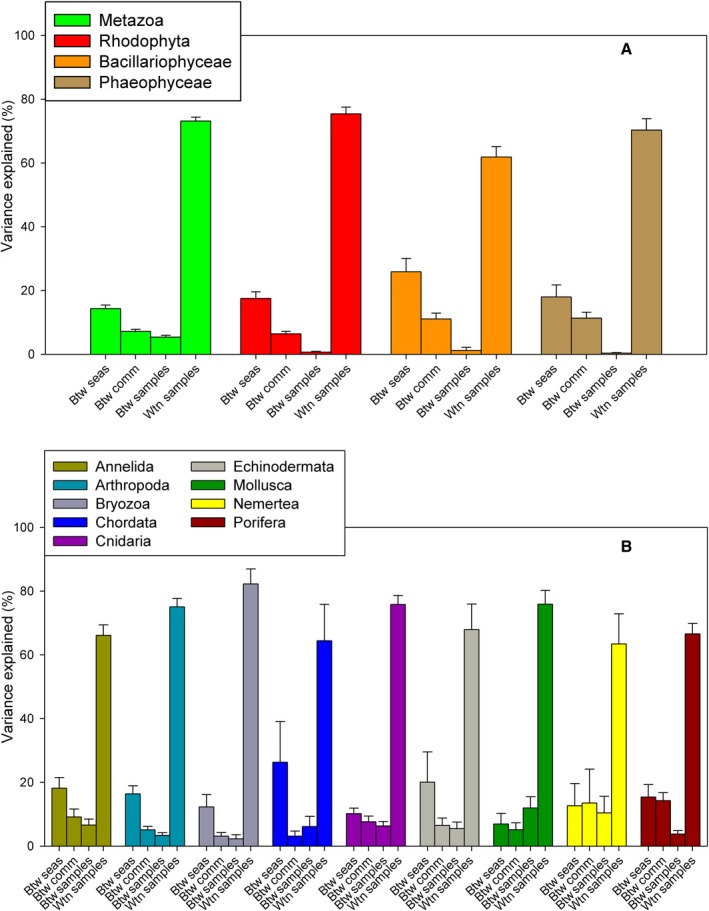
Metabarcoding is by now a well-established method for biodiversity assessment in terrestrial, freshwater, and marine environments. Metabarcoding data sets are usually used for α- and β-diversity estimates, that is, interspecies (or inter-MOTU [molecular operational taxonomic unit]) patterns. However, the use of hypervariable metabarcoding markers may provide an enormous amount of intraspecies (intra-MOTU) information-mostly untapped so far. The use of cytochrome oxidase (COI) amplicons is gaining momentum in metabarcoding studies targeting eukaryote richness. COI has been for a long time the marker of choice in population genetics and phylogeographic studies. Therefore, COI metabarcoding data sets may be used to study intraspecies patterns and phylogeographic features for hundreds of species simultaneously, opening a new field that we suggest to name metaphylogeography. The main challenge for the implementation of this approach is the separation of erroneous sequences from true intra-MOTU variation. Here, we develop a cleaning protocol based on changes in entropy of the different codon positions of the COI sequence, together with co-occurrence patterns of sequences. Using a data set of community DNA from several benthic littoral communities in the Mediterranean and Atlantic seas, we first tested by simulation on a subset of sequences a two-step cleaning approach consisting of a denoising step followed by a minimal abundance filtering. The procedure was then applied to the whole data set. We obtained a total of 563 MOTUs that were usable for phylogeographic inference. We used semiquantitative rank data instead of read abundances to perform AMOVAs and haplotype networks. Genetic variability was mainly concentrated within samples, but with an important between seas component as well. There were intergroup differences in the amount of variability between and within communities in each sea. For two species, the results could be compared with traditional Sanger sequence data available for the same zones, giving similar patterns. Our study shows that metabarcoding data can be used to infer intra- and interpopulation genetic variability of many species at a time, providing a new method with great potential for basic biogeography, connectivity and dispersal studies, and for the more applied fields of conservation genetics, invasion genetics, and design of protected areas.
现在,代谢条形码技术已成为陆地、淡水和海洋环境生物多样性评估的一种成熟方法。代谢条形码数据集通常用于 α-和 β-多样性估计,即种间(或 MOTU 间)模式。然而,超变异代谢条形码标记的使用可能会提供大量的种内(MOTU 内)信息——迄今为止,这些信息大多尚未被挖掘。在针对真核生物丰富度的代谢条形码研究中,细胞色素氧化酶(COI)扩增子的使用正在获得动力。COI 长期以来一直是种群遗传学和系统地理学研究的首选标记。因此,COI 代谢条形码数据集可用于同时研究数百个物种的种内模式和系统地理学特征,开辟了一个我们建议命名为“元系统地理学”的新领域。实施这种方法的主要挑战是将错误序列与真正的 MOTU 内变异区分开来。在这里,我们基于 COI 序列不同密码子位置的熵变化以及序列的共现模式,开发了一种清理协议。我们使用来自地中海和大西洋几个滨岸浅海群落的群落 DNA 数据集,首先在序列子集上通过模拟测试了一种两步清理方法,该方法由去噪步骤和最小丰度过滤组成。然后将该程序应用于整个数据集。我们总共获得了 563 个 MOTU,可用于系统地理学推断。我们使用半定量等级数据而不是读取丰度来执行 AMOVA 和单倍型网络分析。遗传变异性主要集中在样本内,但也有重要的海洋间成分。两个海洋中每个群落内和群落间的变异性都存在组间差异。对于两个物种,结果可以与同一区域可用的传统 Sanger 序列数据进行比较,得到相似的模式。我们的研究表明,代谢条形码数据可用于同时推断许多物种的种内和种群遗传变异性,为基础生物地理学、连通性和扩散研究以及保护遗传学、入侵遗传学和保护区设计等更应用领域提供了一种具有巨大潜力的新方法。