College of Animal Science and Technology, China Agricultural University, Beijing, 100193, China.
USDA-ARS, Animal Genomics and Improvement Laboratory, Beltsville, MD, 20705, USA.
BMC Genomics. 2019 Nov 21;20(1):888. doi: 10.1186/s12864-019-6228-6.
DNA methylation has been shown to be involved in many biological processes, including X chromosome inactivation in females, paternal genomic imprinting, and others.
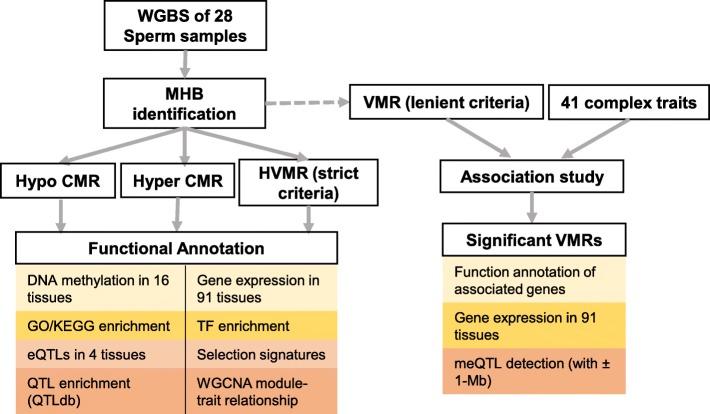
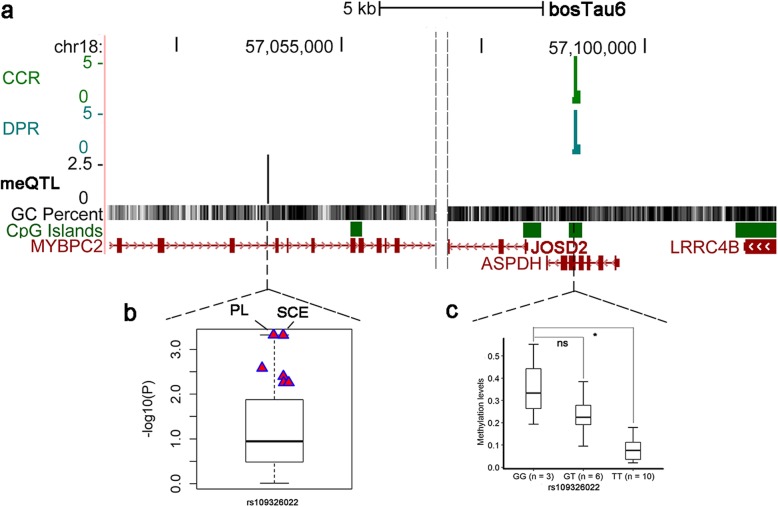
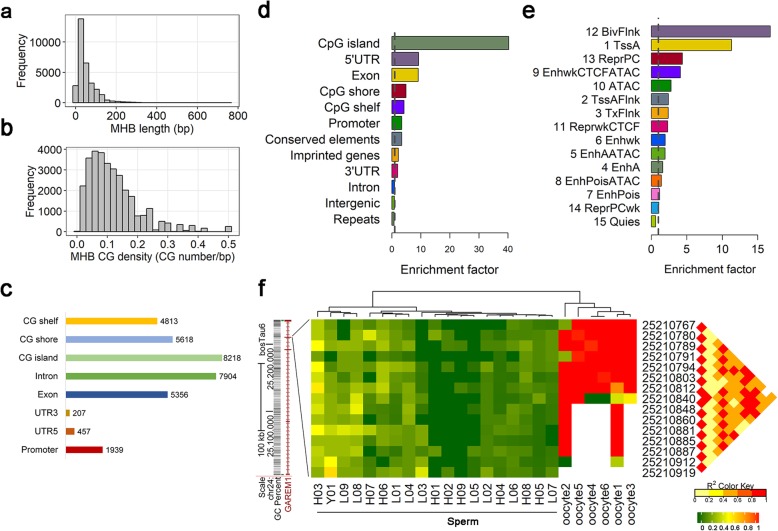
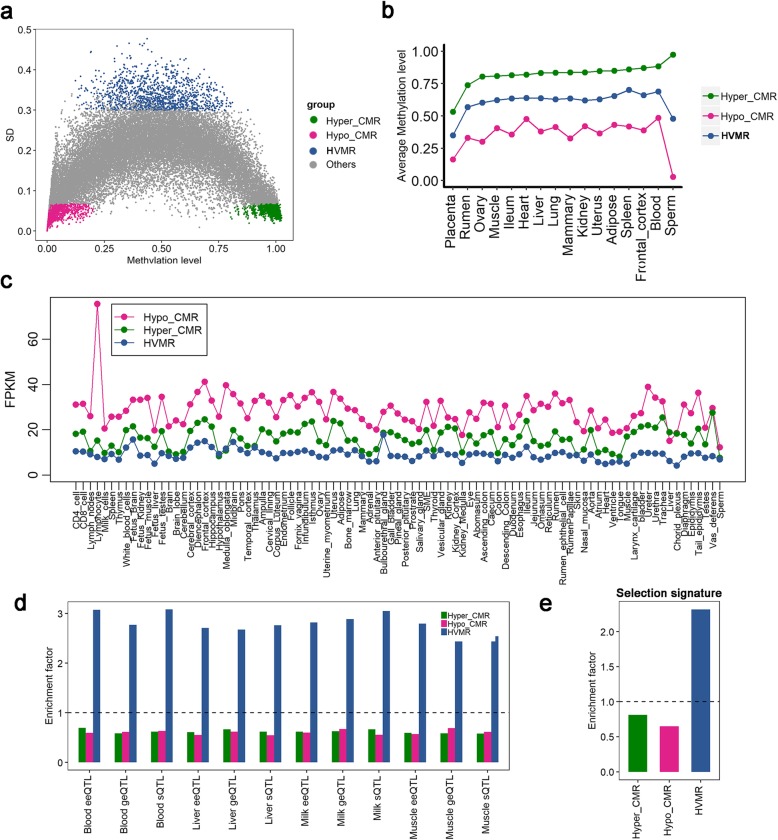
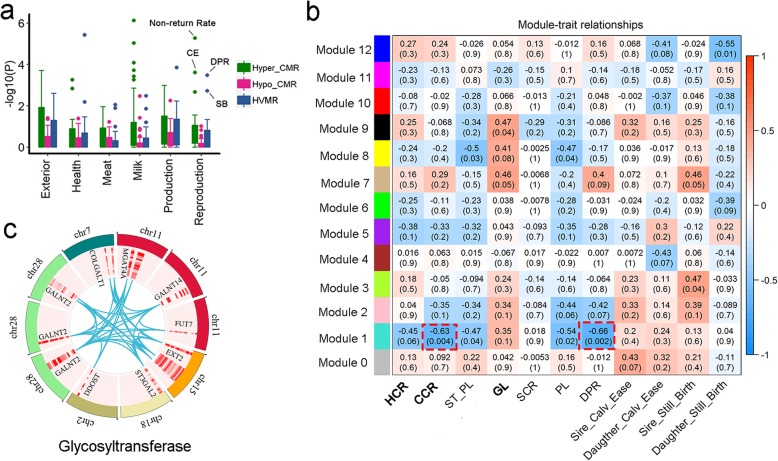
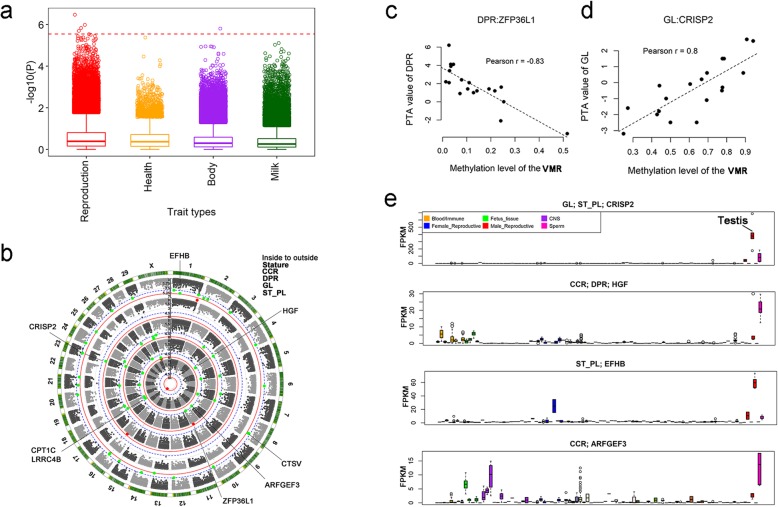
Based on the correlation patterns of methylation levels of neighboring CpG sites among 28 sperm whole genome bisulfite sequencing (WGBS) data (486 × coverage), we obtained 31,272 methylation haplotype blocks (MHBs). Among them, we defined conserved methylated regions (CMRs), variably methylated regions (VMRs) and highly variably methylated regions (HVMRs) among individuals, and showed that HVMRs might play roles in transcriptional regulation and function in complex traits variation and adaptive evolution by integrating evidence from traditional and molecular quantitative trait loci (QTL), and selection signatures. Using a weighted correlation network analysis (WGCNA), we also detected a co-regulated module of HVMRs that was significantly associated with reproduction traits, and enriched for glycosyltransferase genes, which play critical roles in spermatogenesis and fertilization. Additionally, we identified 46 VMRs significantly associated with reproduction traits, nine of which were regulated by cis-SNPs, implying the possible intrinsic relationships among genomic variations, DNA methylation, and phenotypes. These significant VMRs were co-localized (± 10 kb) with genes related to sperm motility and reproduction, including ZFP36L1, CRISP2 and HGF. We provided further evidence that rs109326022 within a predominant QTL on BTA18 might influence the reproduction traits through regulating the methylation level of nearby genes JOSD2 and ASPDH in sperm.
In summary, our results demonstrated associations of sperm DNA methylation with reproduction traits, highlighting the potential of epigenomic information in genomic improvement programs for cattle.
DNA 甲基化参与许多生物学过程,包括女性 X 染色体失活、父本基因组印迹等。
基于 28 个精子全基因组亚硫酸氢盐测序(WGBS)数据(486×覆盖度)中相邻 CpG 位点甲基化水平的相关模式,我们获得了 31272 个甲基化单倍型块(MHB)。其中,我们定义了个体间保守的甲基化区域(CMRs)、可变甲基化区域(VMRs)和高度可变甲基化区域(HVMRs),并通过整合传统和分子数量性状位点(QTL)以及选择信号的证据表明,HVMRs 可能在转录调控和复杂性状变异及适应性进化中发挥作用。使用加权相关网络分析(WGCNA),我们还检测到 HVMRs 的一个共同调控模块,该模块与生殖性状显著相关,并富含糖苷转移酶基因,这些基因在精子发生和受精中起着关键作用。此外,我们还鉴定出 46 个与生殖性状显著相关的 VMR,其中 9 个受顺式-SNPs 调控,这暗示了基因组变异、DNA 甲基化和表型之间可能存在内在关系。这些显著的 VMR 与精子运动和生殖相关基因共定位(±10kb),包括 ZFP36L1、CRISP2 和 HGF。我们提供了进一步的证据表明,BTA18 上主要 QTL 内的 rs109326022 可能通过调节精子中附近基因 JOSD2 和 ASPDH 的甲基化水平来影响生殖性状。
总之,我们的研究结果表明精子 DNA 甲基化与生殖性状之间存在关联,突出了表观基因组信息在牛基因组改良计划中的潜力。