Milko Laura V, Chen Flavia, Chan Kee, Brower Amy M, Agrawal Pankaj B, Beggs Alan H, Berg Jonathan S, Brenner Steven E, Holm Ingrid A, Koenig Barbara A, Parad Richard B, Powell Cynthia M, Kingsmore Stephen F
1Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599 USA.
2Institute for Human Genetics, University of California, San Francisco, CA 94143 USA.
NPJ Genom Med. 2019 Dec 10;4:32. doi: 10.1038/s41525-019-0105-8. eCollection 2019.
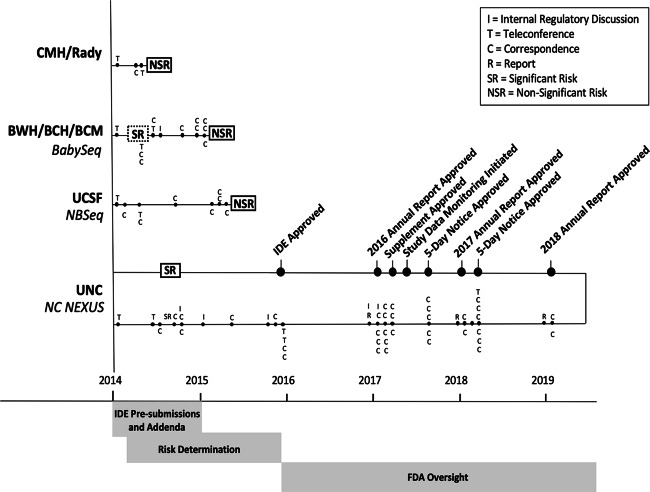
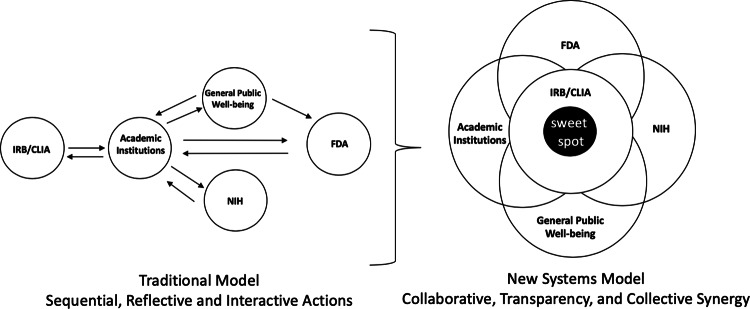
The National Institutes of Health (NIH) funded the Newborn Sequencing In Genomic medicine and public HealTh (NSIGHT) Consortium to investigate the implications, challenges, and opportunities associated with the possible use of genomic sequence information in the newborn period. Following announcement of the NSIGHT awardees in 2013, the Food and Drug Administration (FDA) contacted investigators and requested that pre-submissions to investigational device exemptions (IDE) be submitted for the use of genomic sequencing under Title 21 of the Code of Federal Regulations (21 CFR) part 812. IDE regulation permits clinical investigation of medical devices that have not been approved by the FDA. To our knowledge, this marked the first time the FDA determined that NIH-funded clinical genomic research projects are subject to IDE regulation. Here, we review the history of and rationale behind FDA oversight of clinical research and the NSIGHT Consortium's experiences in navigating the IDE process. Overall, NSIGHT investigators found that FDA's application of existing IDE regulations and medical device definitions aligned imprecisely with the aims of publicly funded exploratory clinical research protocols. IDE risk assessments by the FDA were similar to, but distinct from, protocol risk assessments conducted by local Institutional Review Boards (IRBs), and had the potential to reflect novel oversight of emerging genomic technologies. However, the pre-IDE and IDE process delayed the start of NSIGHT research studies by an average of 10 months, and significantly limited the scope of investigation in two of the four NIH approved projects. Based on the experience of the NSIGHT Consortium, we conclude that policies and practices governing the development and use of novel genomic technologies in clinical research urgently need clarification in order to mitigate potentially conflicting or redundant oversight by IRBs, NIH, FDA, and state authorities.
美国国立卫生研究院(NIH)资助了基因组医学与公共卫生新生儿测序(NSIGHT)联盟,以研究在新生儿期使用基因组序列信息可能带来的影响、挑战和机遇。2013年NSIGHT获奖者名单公布后,美国食品药品监督管理局(FDA)联系了研究人员,并要求根据联邦法规法典(CFR)第21编第812部分提交关于使用基因组测序的研究性器械豁免(IDE)预提交文件。IDE法规允许对未经FDA批准的医疗器械进行临床研究。据我们所知,这是FDA首次认定由NIH资助的临床基因组研究项目受IDE法规监管。在此,我们回顾FDA对临床研究进行监督的历史和依据,以及NSIGHT联盟在IDE流程中的经历。总体而言,NSIGHT研究人员发现,FDA对现有IDE法规和医疗器械定义的应用与公共资助的探索性临床研究方案的目标并不完全一致。FDA进行的IDE风险评估与当地机构审查委员会(IRB)进行的方案风险评估相似但不同,并且有可能反映对新兴基因组技术的新监督。然而,IDE预提交和IDE流程使NSIGHT研究项目的启动平均推迟了10个月,并显著限制了NIH批准的四个项目中两个项目的研究范围。基于NSIGHT联盟的经验,我们得出结论,迫切需要澄清临床研究中新型基因组技术开发和使用的政策与实践,以减轻IRB、NIH、FDA和州当局潜在的相互冲突或冗余的监督。