Shen Nan, Du Jun, Zhou Hui, Chen Nan, Pan Yi, Hoheisel Jörg D, Jiang Zonghui, Xiao Ling, Tao Yue, Mo Xi
Department of Infectious Diseases, Shanghai Children's Medical Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Pediatric Translational Medicine Institute, Shanghai Children's Medical Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Front Oncol. 2019 Dec 3;9:1281. doi: 10.3389/fonc.2019.01281. eCollection 2019.
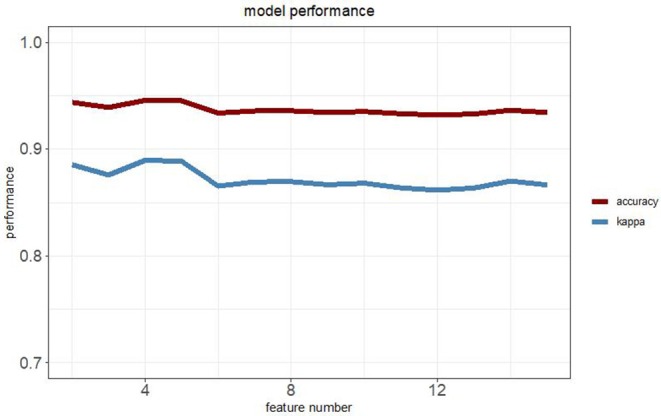
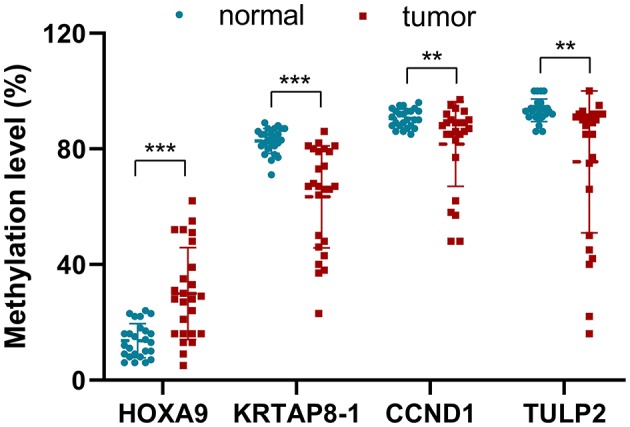
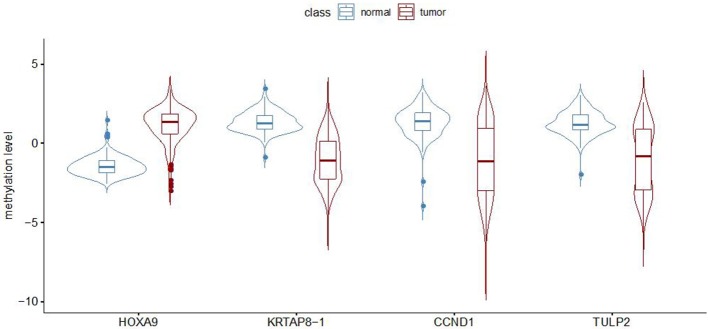
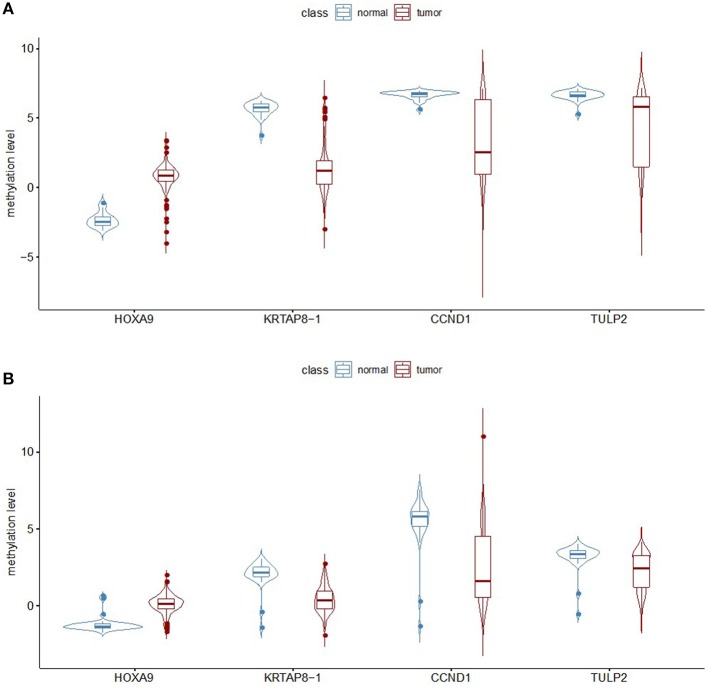
Lung adenocarcinoma (LUAD) is one of the most common cancers and lethal diseases in the world. Recognition of the undetermined lung nodules at an early stage is useful for a favorable prognosis. However, there is no good method to identify the undetermined lung nodules and predict their clinical outcome. DNA methylation alteration is frequently observed in LUAD and may play important roles in carcinogenesis, diagnosis, and prediction. This study took advantage of publicly available methylation profiling resources and a machine learning method to investigate methylation differences between LUAD and adjacent non-malignant tissue. The prediction panel was first constructed using 338 tissue samples from LUAD patients including 149 non-malignant ones. This model was then validated with data from The Cancer Genome Atlas database and clinic samples. As a result, the methylation status of four CpG loci in homeobox A9 (), keratin-associated protein 8-1 (), cyclin D1 (), and tubby-like protein 2 () were highlighted as informative markers. A random forest classification model with an accuracy of 94.57% and kappa of 88.96% was obtained. To evaluate this panel for LUAD, the methylation levels of four CpG loci in , and of tumor samples and matched adjacent lung samples from 25 patients with LUAD were tested. In these LUAD patients, the methylation of was significantly upregulated, whereas the methylation of , and were downregulated obviously in tumor samples compared with adjacent tissues. Our study demonstrates that the methylation of , and has great potential for the early recognition of LUAD in the undetermined lung nodules. The findings also exhibit that the application of improved mathematic algorithms can yield accurate and particularly robust and widely applicable marker panels. This approach could greatly facilitate the discovery process of biomarkers in various fields.
肺腺癌(LUAD)是世界上最常见的癌症和致命疾病之一。早期识别未确定的肺结节对良好的预后有益。然而,目前尚无识别未确定肺结节并预测其临床结果的有效方法。DNA甲基化改变在肺腺癌中经常被观察到,可能在致癌、诊断和预测中发挥重要作用。本研究利用公开可用的甲基化谱资源和机器学习方法,研究肺腺癌与相邻非恶性组织之间的甲基化差异。首先使用来自149例非恶性肺腺癌患者的338个组织样本构建预测模型。然后用来自癌症基因组图谱数据库的数据和临床样本对该模型进行验证。结果,同源框A9()、角蛋白相关蛋白8-1()、细胞周期蛋白D1()和类 Tubby 蛋白2()中四个CpG位点的甲基化状态被确定为信息性标志物。获得了一个随机森林分类模型,其准确率为94.57%,kappa值为88.96%。为了评估该模型对肺腺癌的诊断价值,检测了25例肺腺癌患者肿瘤样本及配对的相邻肺组织样本中、和四个CpG位点的甲基化水平。在这些肺腺癌患者中,肿瘤样本中的甲基化显著上调,而与相邻组织相比,、和的甲基化明显下调。我们的研究表明,、和的甲基化在早期识别未确定肺结节中的肺腺癌具有巨大潜力。研究结果还表明,改进的数学算法的应用可以产生准确、特别稳健且广泛适用标记物模型。这种方法可以极大地促进各个领域生物标志物的发现过程。