Institute of Chemical Biology and Fundamental Medicine SB RAS, Lavrent'ev Ave, 8, 630090 Novosibirsk, Russia.
Federal Research Centre, Institute of Cytology and Genetics SB RAS, Lavrent'ev Ave, 10, 630090 Novosibirsk, Russia.
Int J Mol Sci. 2019 Dec 27;21(1):214. doi: 10.3390/ijms21010214.
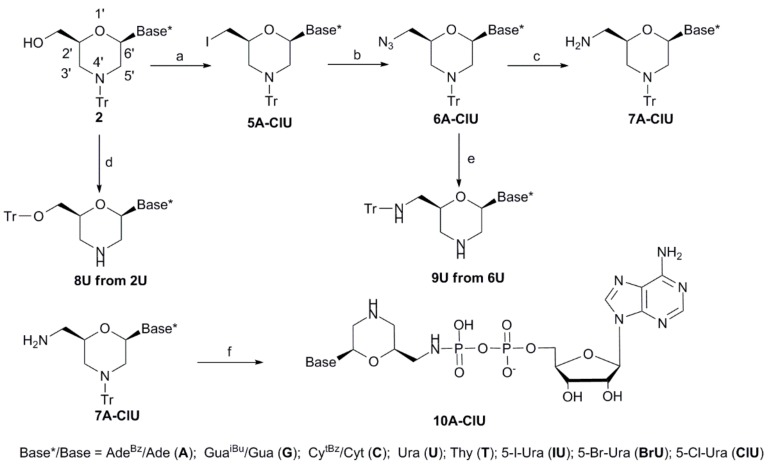
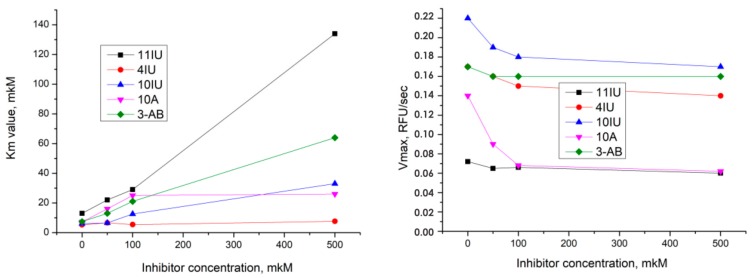
We report on the design, synthesis and molecular modeling study of conjugates of adenosine diphosphate (ADP) and morpholino nucleosides as potential selective inhibitors of poly(ADP-ribose)polymerases-1, 2 and 3. Sixteen dinucleoside pyrophosphates containing natural heterocyclic bases as well as 5-haloganeted pyrimidines, and mimicking a main substrate of these enzymes, nicotinamide adenine dinucleotide (NAD+)-molecule, have been synthesized in a high yield. Morpholino nucleosides have been tethered to the β-phosphate of ADP via a phosphoester or phosphoramide bond. Screening of the inhibiting properties of these derivatives on the autopoly(ADP-ribosyl)ation of PARP-1 and PARP-2 has shown that the effect depends upon the type of nucleobase as well as on the linkage between ADP and morpholino nucleoside. The 5-iodination of uracil and the introduction of the P-N bond in NAD+-mimetics have shown to increase inhibition properties. Structural modeling suggested that the P-N bond can stabilize the pyrophosphate group in active conformation due to the formation of an intramolecular hydrogen bond. The most active NAD+ analog against PARP-1 contained 5-iodouracil 2'-aminomethylmorpholino nucleoside with IC50 126 ± 6 μM, while in the case of PARP-2 it was adenine 2'-aminomethylmorpholino nucleoside (IC50 63 ± 10 μM). In silico analysis revealed that thymine and uracil-based NAD+ analogs were recognized as the NAD+-analog that targets the nicotinamide binding site. On the contrary, the adenine 2'-aminomethylmorpholino nucleoside-based NAD+ analogs were predicted to identify as PAR-analogs that target the acceptor binding site of PARP-2, representing a novel molecular mechanism for selective PARP inhibition. This discovery opens a new avenue for the rational design of PARP-1/2 specific inhibitors.
我们报告了腺嘌呤二核苷酸(ADP)和吗啉核苷缀合物的设计、合成和分子建模研究,这些缀合物作为聚(ADP-核糖)聚合酶-1、2 和 3 的潜在选择性抑制剂。十六个包含天然杂环碱基的二核苷焦磷酸盐以及模拟这些酶的主要底物烟酰胺腺嘌呤二核苷酸(NAD+)分子的 5-卤代嘧啶已通过高收率合成。通过磷酸酯或磷酰胺键将吗啉核苷连接到 ADP 的 β-磷酸上。对这些衍生物对 PARP-1 和 PARP-2 自身多聚(ADP-核糖基)化的抑制特性进行筛选表明,这种作用取决于碱基的类型以及 ADP 和吗啉核苷之间的连接。尿嘧啶的 5-碘化和 NAD+模拟物中 P-N 键的引入表明抑制特性增加。结构建模表明,P-N 键可以通过形成分子内氢键稳定活性构象中的焦磷酸盐基团。对 PARP-1 最有效的 NAD+类似物含有 5-碘尿嘧啶 2'-氨甲基吗啉核苷,IC50 为 126 ± 6 μM,而对 PARP-2 则为腺嘌呤 2'-氨甲基吗啉核苷(IC50 为 63 ± 10 μM)。计算机分析表明,胸腺嘧啶和尿嘧啶基 NAD+类似物被识别为靶向烟酰胺结合位点的 NAD+-类似物。相反,预测腺嘌呤 2'-氨甲基吗啉核苷基 NAD+类似物被鉴定为靶向 PARP-2 受体结合位点的 PAR 类似物,代表了 PARP 抑制的新分子机制。这一发现为 PARP-1/2 特异性抑制剂的合理设计开辟了新途径。