Department of Clinical Chinese Pharmacy, School of Chinese Materia Medica, Beijing University of Chinese Medicine, Beijing, China (mainland).
Evidence Based Medicine Center, School of Basic Medical Science, Lanzhou University, Lanzhou, Gansu, China (mainland).
Med Sci Monit. 2020 Jan 1;26:e918906. doi: 10.12659/MSM.918906.
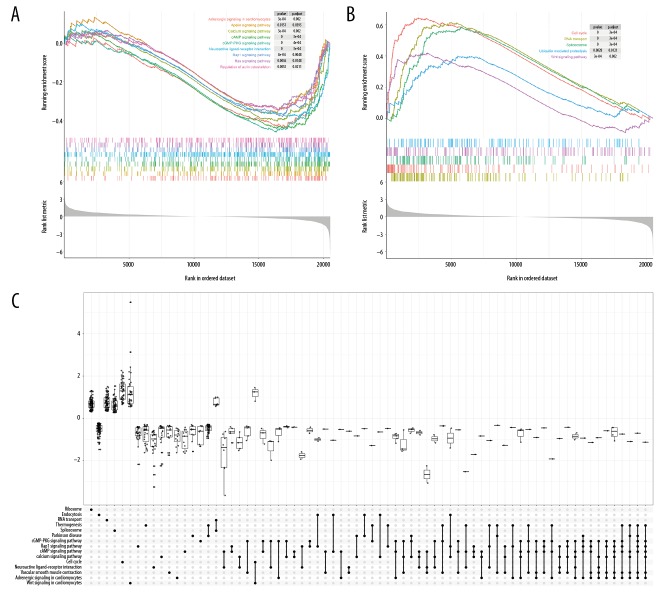
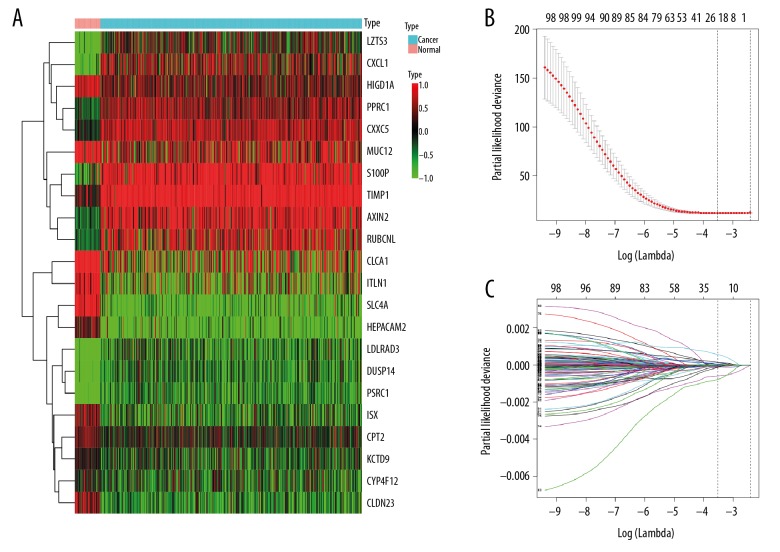
BACKGROUND Despite noteworthy advancements in the multidisciplinary treatment of colorectal cancer (CRC) and deeper understanding in the molecular mechanisms of CRC, many of CRC patients with histologically identical tumors present different treatment response and prognosis. Thus, more evidence on novel predictive and prognostic biomarkers for CRC remains urgently needed. This study aims to identify potential prognostic biomarkers for CRC with integrative gene expression profiling analysis. MATERIAL AND METHODS Differential expression analysis of paired CRC and adjacent normal tissue samples in 6 microarray datasets was independently performed, and the 6 datasets were integrated by the robust rank aggregation method to detect consistent differentially expressed genes (DEGs). Aberrant expression patterns of these genes were further validated in RNA sequencing data. Then, gene set enrichment analysis (GSEA) was performed to investigate significantly dysregulated biological functions in CRC. Finally, univariate, LASSO and multivariate Cox regression models were built to identify key prognostic genes in CRC patients. RESULTS A total of 990 DEGs (495 downregulated and 495 upregulated genes) were acquired after integratedly analyzing the 6 microarray datasets, and 4131 DEGs (2050 downregulated and 2081 upregulated genes) were obtained from the RNA sequencing dataset. Subsequently, these DEGs were intersected and 885 consistent DEGs were finally identified, including 458 downregulated and 427 upregulated genes. Two risky prognostic genes (TIMP1 and LZTS3) and 5 protective prognostic genes (AXIN2, CXCL1, ITLN1, CPT2 and CLDN23) were identified, which were significantly associated with the prognosis of CRC. CONCLUSIONS The 7 genes that we identified would provide more evidence for further applying novel diagnostic and prognostic biomarkers in clinical practice to facilitate personalized treatment of CRC.
尽管结直肠癌(CRC)的多学科治疗取得了显著进展,对 CRC 分子机制的理解也更加深入,但许多组织学上相同肿瘤的 CRC 患者表现出不同的治疗反应和预后。因此,迫切需要更多关于 CRC 新型预测和预后生物标志物的证据。本研究旨在通过整合基因表达谱分析鉴定 CRC 的潜在预后生物标志物。
对 6 个微阵列数据集的配对 CRC 和相邻正常组织样本进行差异表达分析,通过稳健秩聚合方法对 6 个数据集进行整合,以检测一致的差异表达基因(DEGs)。这些基因的异常表达模式在 RNA 测序数据中进一步验证。然后,进行基因集富集分析(GSEA)以研究 CRC 中显著失调的生物学功能。最后,构建单变量、LASSO 和多变量 Cox 回归模型,以识别 CRC 患者的关键预后基因。
综合分析 6 个微阵列数据集后,共获得 990 个 DEGs(495 个下调和 495 个上调基因),从 RNA 测序数据集获得 4131 个 DEGs(2050 个下调和 2081 个上调基因)。随后,这些 DEGs 进行了交集,最终确定了 885 个一致的 DEGs,包括 458 个下调和 427 个上调基因。鉴定出 2 个风险预后基因(TIMP1 和 LZTS3)和 5 个保护预后基因(AXIN2、CXCL1、ITLN1、CPT2 和 CLDN23),它们与 CRC 的预后显著相关。
我们鉴定的这 7 个基因将为进一步在临床实践中应用新型诊断和预后生物标志物提供更多证据,以促进 CRC 的个体化治疗。