Breitbach Megan E, Greenspan Susan, Resnick Neil M, Perera Subashan, Gurkar Aditi U, Absher Devin, Levine Arthur S
HudsonAlpha Institute for Biotechnology, Hunstville, AL, United States.
Department of Biotechnology Science and Engineering, University of Alabama in Huntsville, Hunstville, AL, United States.
Front Genet. 2019 Dec 19;10:1277. doi: 10.3389/fgene.2019.01277. eCollection 2019.
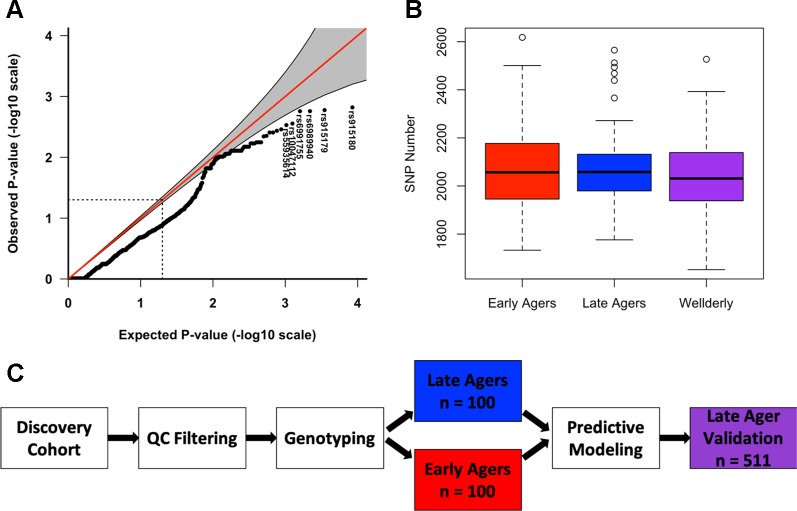
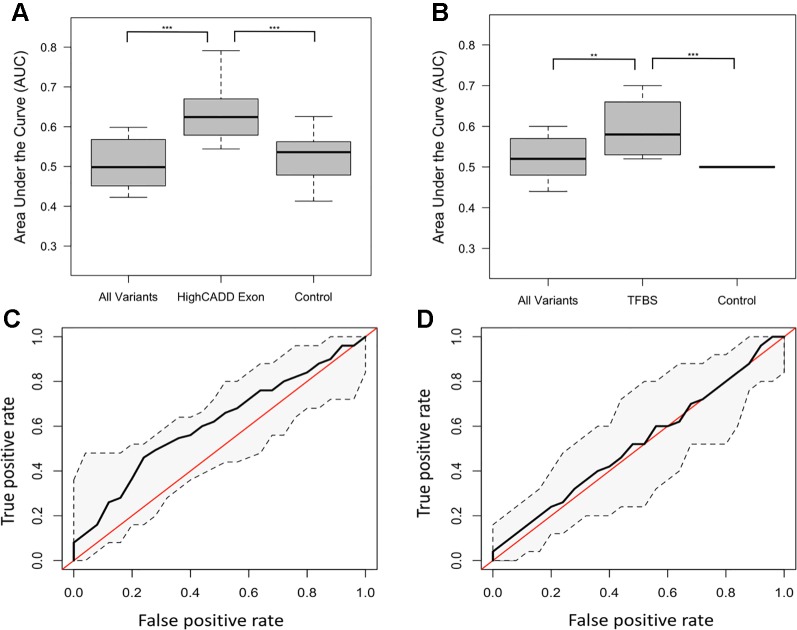
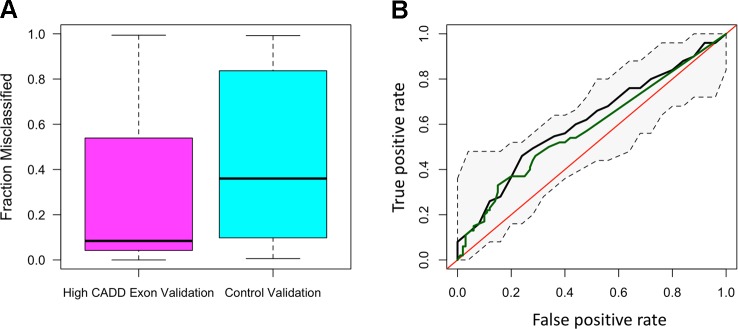
Recent studies investigating longevity have revealed very few convincing genetic associations with increased lifespan. This is, in part, due to the complexity of biological aging, as well as the limited power of genome-wide association studies, which assay common single nucleotide polymorphisms (SNPs) and require several thousand subjects to achieve statistical significance. To overcome such barriers, we performed comprehensive DNA sequencing of a panel of 20 genes previously associated with phenotypic aging in a cohort of 200 individuals, half of whom were clinically defined by an "early aging" phenotype, and half of whom were clinically defined by a "late aging" phenotype based on age (65-75 years) and the ability to walk up a flight of stairs or walk for 15 min without resting. A validation cohort of 511 late agers was used to verify our results. We found early agers were not enriched for more total variants in these 20 aging-related genes than late agers. Using machine learning methods, we identified the most predictive model of aging status, both in our discovery and validation cohorts, to be a random forest model incorporating damaging exon variants [Combined Annotation-Dependent Depletion (CADD) > 15]. The most heavily weighted variants in the model were within () (), both of which are involved in a canonical aging pathway, DNA damage repair. Overall, this study implemented a framework to apply machine learning to identify sequencing variants associated with complex phenotypes such as aging. While the small sample size making up our cohort inhibits our ability to make definitive conclusions about the ability of these genes to accurately predict aging, this study offers a unique method for exploring polygenic associations with complex phenotypes.
近期关于长寿的研究显示,与寿命延长相关的令人信服的基因关联非常少。部分原因在于生物衰老的复杂性,以及全基因组关联研究的效力有限,这类研究检测常见的单核苷酸多态性(SNP),需要数千名受试者才能达到统计学显著性。为克服这些障碍,我们对一组20个先前与表型衰老相关的基因进行了全面的DNA测序,研究对象为200名个体,其中一半根据“早衰”表型进行临床定义,另一半根据年龄(65 - 75岁)以及爬一段楼梯或步行15分钟不休息的能力,按照“晚衰”表型进行临床定义。使用511名晚衰者组成的验证队列来验证我们的结果。我们发现,在这20个与衰老相关的基因中,早衰者并不比晚衰者富集更多的总变异。使用机器学习方法,我们在发现队列和验证队列中均确定,衰老状态的最具预测性模型是一个纳入有害外显子变异(综合注释依赖损耗,CADD>15)的随机森林模型。该模型中权重最大的变异位于()()内,这两个基因均参与一个典型的衰老途径——DNA损伤修复。总体而言,本研究实施了一个框架,应用机器学习来识别与衰老等复杂表型相关的测序变异。虽然我们队列的样本量较小,限制了我们就这些基因准确预测衰老的能力得出明确结论,但本研究提供了一种探索多基因与复杂表型关联的独特方法。