Department of Genetics, UMCG HPC CB50, RB Groningen, Netherlands.
Institute of Ageing and Chronic Disease, University of Liverpool, Liverpool, UK.
Brief Bioinform. 2018 Jul 20;19(4):575-592. doi: 10.1093/bib/bbw139.
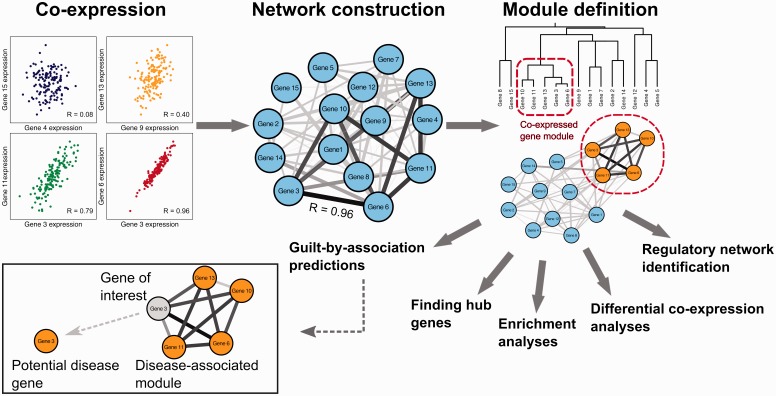
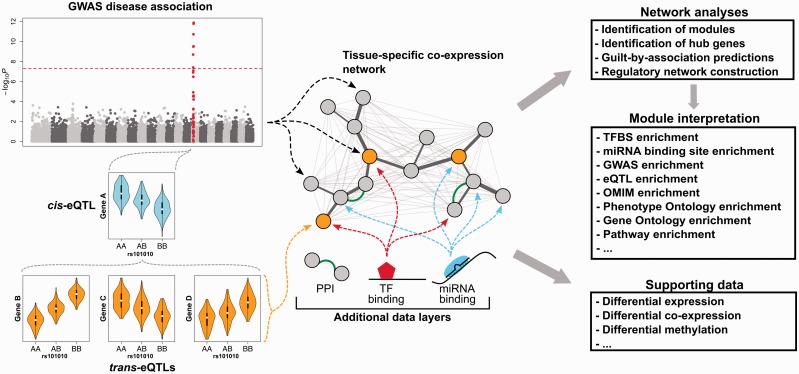
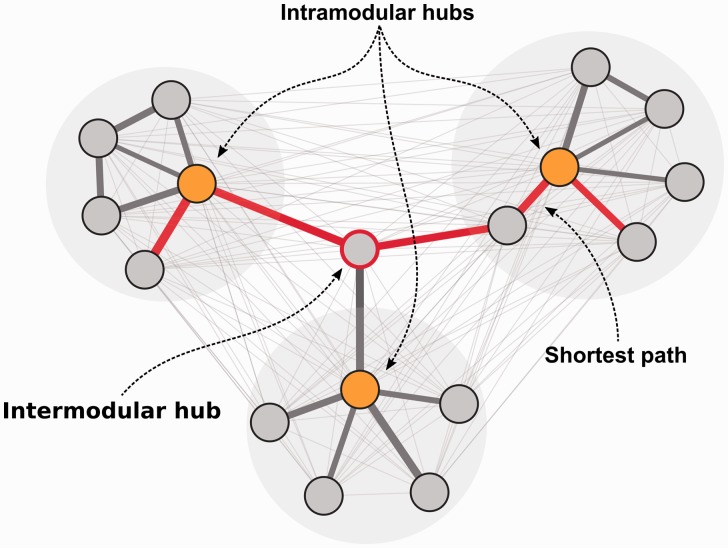
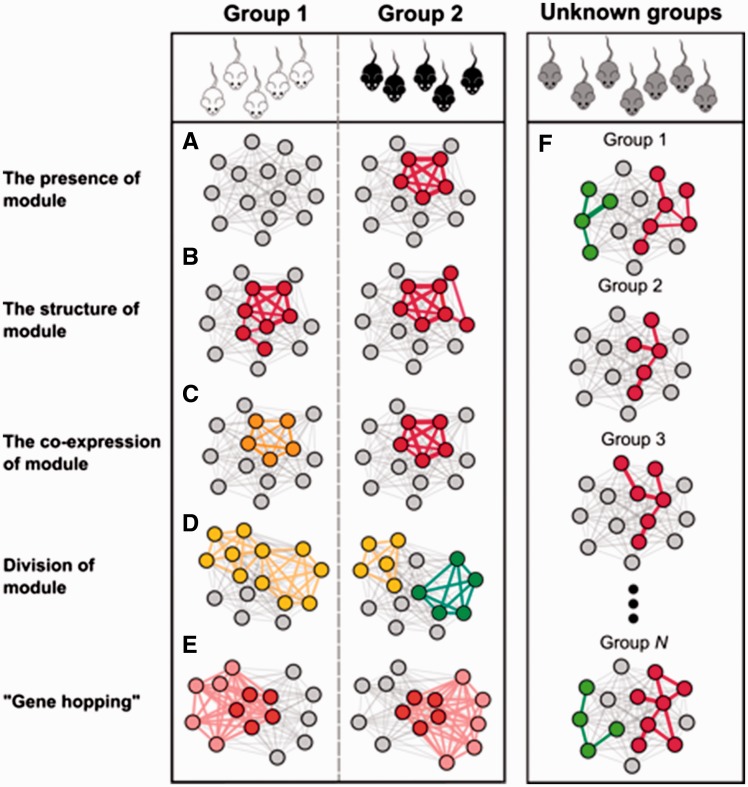
Gene co-expression networks can be used to associate genes of unknown function with biological processes, to prioritize candidate disease genes or to discern transcriptional regulatory programmes. With recent advances in transcriptomics and next-generation sequencing, co-expression networks constructed from RNA sequencing data also enable the inference of functions and disease associations for non-coding genes and splice variants. Although gene co-expression networks typically do not provide information about causality, emerging methods for differential co-expression analysis are enabling the identification of regulatory genes underlying various phenotypes. Here, we introduce and guide researchers through a (differential) co-expression analysis. We provide an overview of methods and tools used to create and analyse co-expression networks constructed from gene expression data, and we explain how these can be used to identify genes with a regulatory role in disease. Furthermore, we discuss the integration of other data types with co-expression networks and offer future perspectives of co-expression analysis.
基因共表达网络可用于将功能未知的基因与生物过程联系起来,优先考虑候选疾病基因或辨别转录调控程序。随着转录组学和下一代测序技术的最新进展,基于 RNA 测序数据构建的共表达网络还可以推断非编码基因和剪接变异体的功能和疾病关联。尽管基因共表达网络通常不提供因果关系的信息,但新兴的差异共表达分析方法正在识别各种表型背后的调节基因。在这里,我们介绍并指导研究人员进行(差异)共表达分析。我们概述了用于创建和分析基于基因表达数据构建的共表达网络的方法和工具,并解释了如何使用这些方法来识别在疾病中具有调节作用的基因。此外,我们还讨论了将其他类型的数据与共表达网络集成,并提供了共表达分析的未来展望。