Wellcome Centre for Human Genetics, Oxford, United Kingdom.
Oxford Centre for Diabetes, Endocrinology and Metabolism, University of Oxford, Oxford, United Kingdom.
Elife. 2020 Jan 27;9:e51503. doi: 10.7554/eLife.51503.
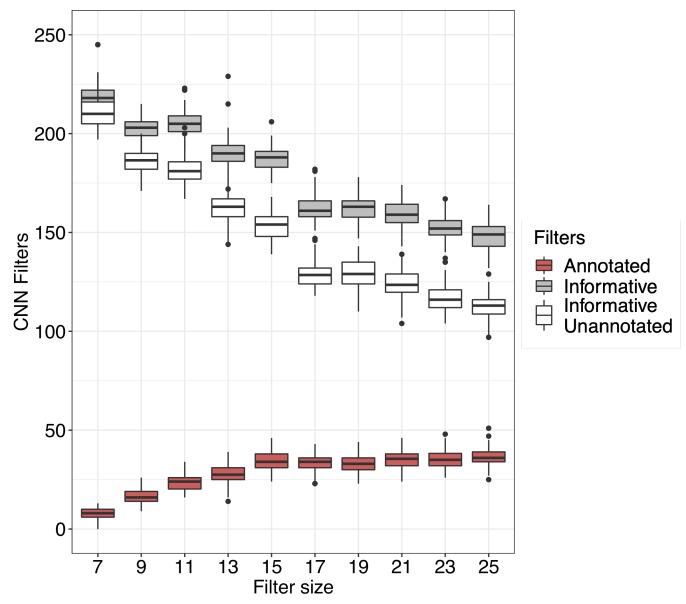
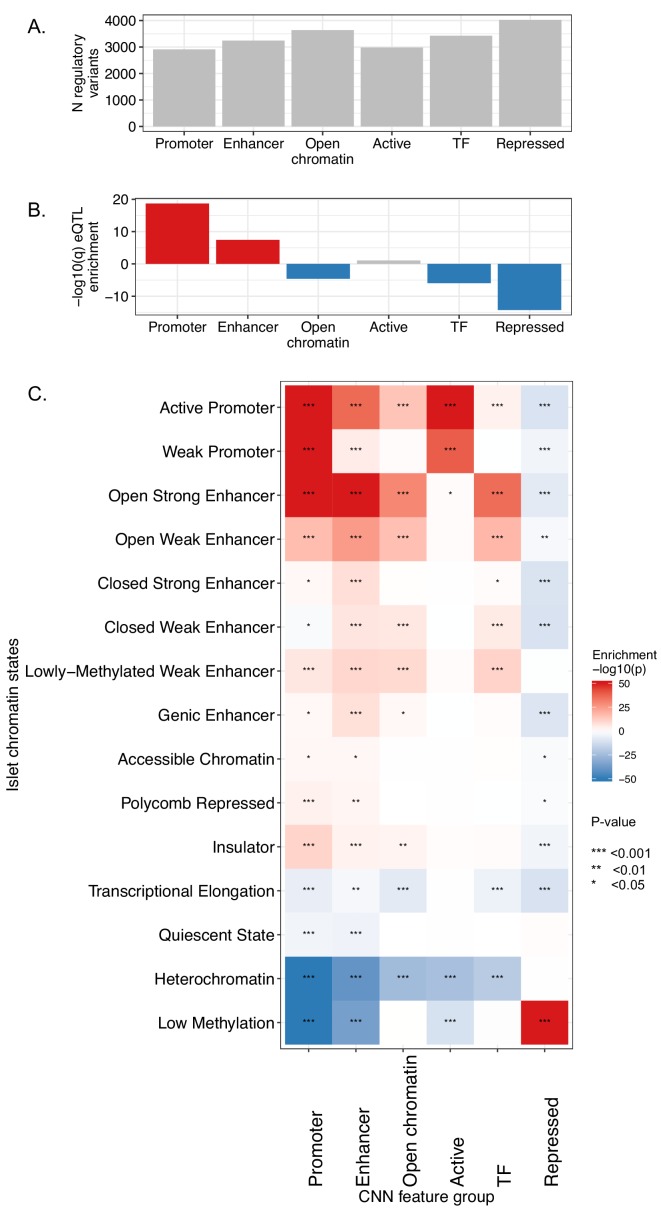
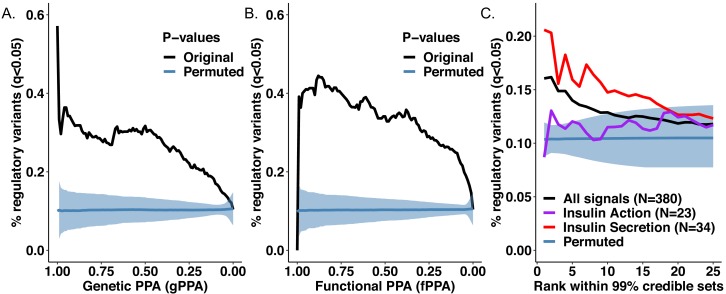
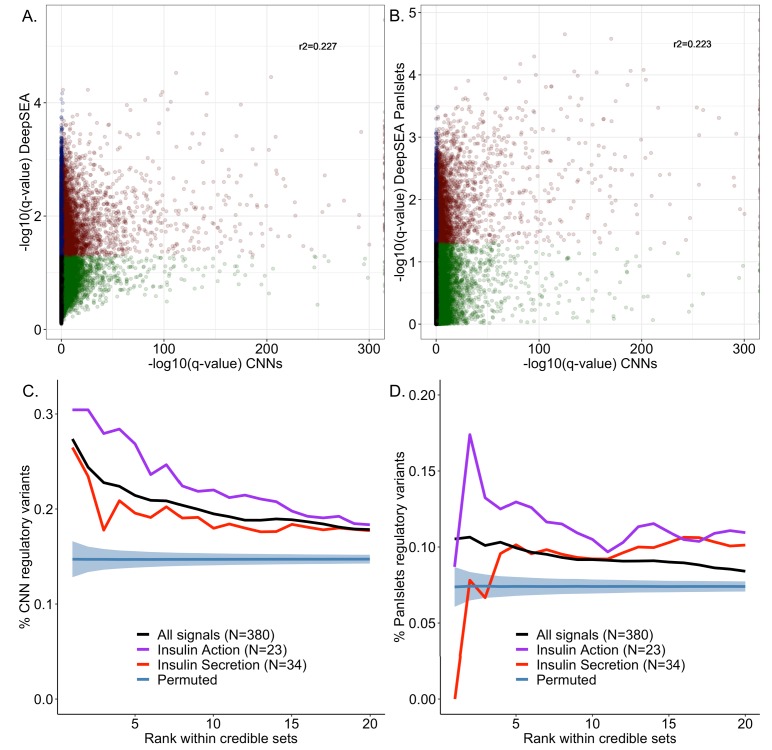
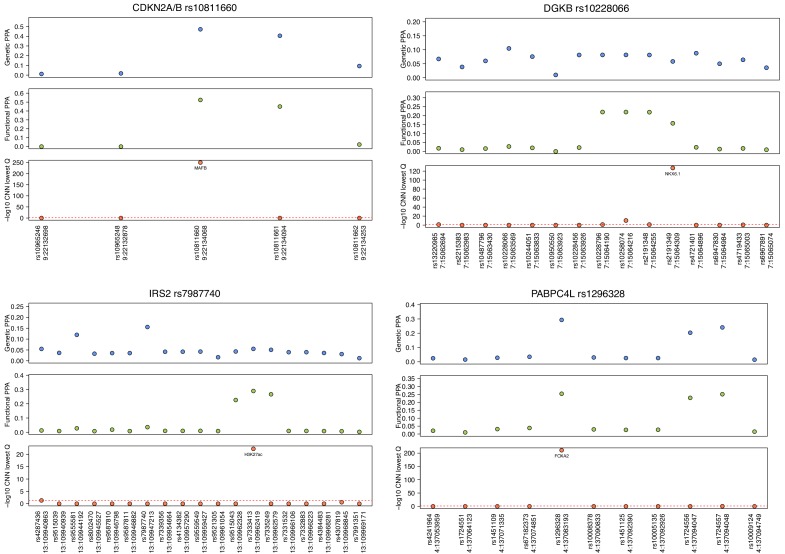
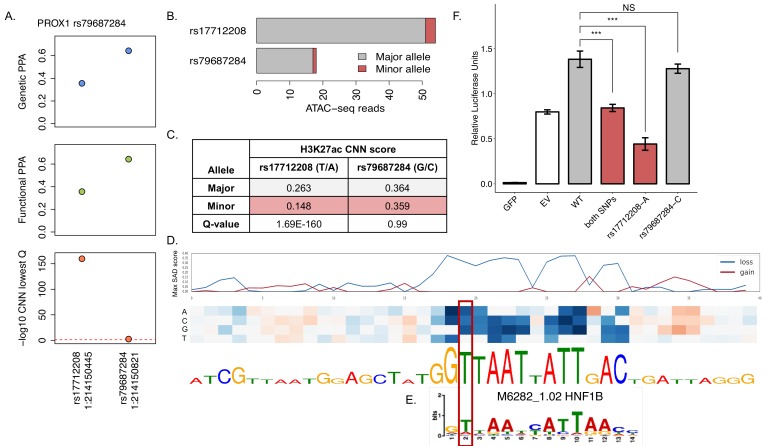
Genome-wide association analyses have uncovered multiple genomic regions associated with T2D, but identification of the causal variants at these remains a challenge. There is growing interest in the potential of deep learning models - which predict epigenome features from DNA sequence - to support inference concerning the regulatory effects of disease-associated variants. Here, we evaluate the advantages of training convolutional neural network (CNN) models on a broad set of epigenomic features collected in a single disease-relevant tissue - pancreatic islets in the case of type 2 diabetes (T2D) - as opposed to models trained on multiple human tissues. We report convergence of CNN-based metrics of regulatory function with conventional approaches to variant prioritization - genetic fine-mapping and regulatory annotation enrichment. We demonstrate that CNN-based analyses can refine association signals at T2D-associated loci and provide experimental validation for one such signal. We anticipate that these approaches will become routine in downstream analyses of GWAS.
全基因组关联分析已经揭示了多个与 T2D 相关的基因组区域,但鉴定这些区域中的因果变异仍然是一个挑战。深度学习模型在预测 DNA 序列中的表观基因组特征方面具有很大的潜力,这些模型可以支持推断与疾病相关变异的调控效应,这引起了越来越多的关注。在这里,我们评估了在单一与疾病相关的组织(即 2 型糖尿病(T2D)的胰岛)中收集的广泛的表观基因组特征上训练卷积神经网络(CNN)模型的优势,而不是在多个人体组织上训练模型。我们报告了基于 CNN 的调控功能指标与传统的变异优先级排序方法(遗传精细映射和调控注释富集)的收敛性。我们证明,基于 CNN 的分析可以细化 T2D 相关基因座的关联信号,并为其中一个信号提供实验验证。我们预计这些方法将成为 GWAS 下游分析的常规方法。