School of Population and Public Health, University of British Columbia, Vancouver, Canada.
Yukon Communicable Disease Control, Health and Social Services, Government of Yukon, Whitehorse, Canada.
Epidemiol Infect. 2020 Feb 4;148:e15. doi: 10.1017/S0950268820000072.
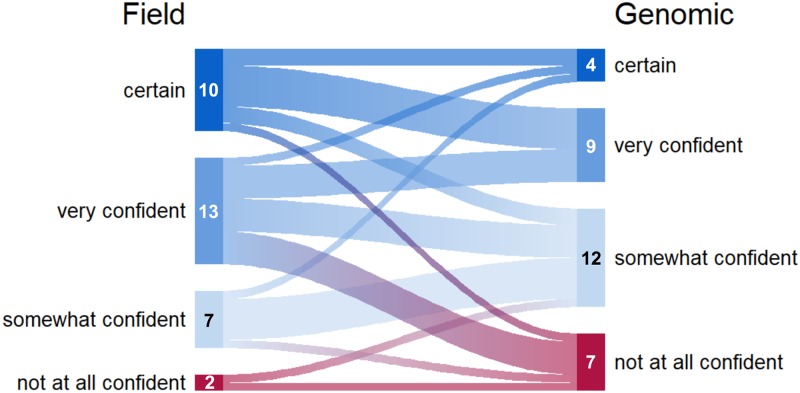
Yukon Territory (YT) is a remote region in northern Canada with ongoing spread of tuberculosis (TB). To explore the utility of whole genome sequencing (WGS) for TB surveillance and monitoring in a setting with detailed contact tracing and interview data, we used a mixed-methods approach. Our analysis included all culture-confirmed cases in YT (2005-2014) and incorporated data from 24-locus Mycobacterial Interspersed Repetitive Units-Variable Number of Tandem Repeats (MIRU-VNTR) genotyping, WGS and contact tracing. We compared field-based (contact investigation (CI) data + MIRU-VNTR) and genomic-based (WGS + MIRU-VNTR + basic case data) investigations to identify the most likely source of each person's TB and assessed the knowledge, attitudes and practices of programme personnel around genotyping and genomics using online, multiple-choice surveys (n = 4) and an in-person group interview (n = 5). Field- and genomics-based approaches agreed for 26 of 32 (81%) cases on likely location of TB acquisition. There was less agreement in the identification of specific source cases (13/22 or 59% of cases). Single-locus MIRU-VNTR variants and limited genetic diversity complicated the analysis. Qualitative data indicated that participants viewed genomic epidemiology as a useful tool to streamline investigations, particularly in differentiating latent TB reactivation from the recent transmission. Based on this, genomic data could be used to enhance CIs, focus resources, target interventions and aid in TB programme evaluation.
育空地区(YT)是加拿大北部的一个偏远地区,结核病(TB)持续蔓延。为了探索全基因组测序(WGS)在一个具有详细接触者追踪和访谈数据的环境中用于结核病监测和监测的实用性,我们采用了混合方法。我们的分析包括育空地区所有培养确诊的病例(2005-2014 年),并纳入了 24 位位点分枝杆菌间隔重复单元-可变数量串联重复(MIRU-VNTR)基因分型、WGS 和接触者追踪的数据。我们比较了基于现场(接触调查(CI)数据+MIRU-VNTR)和基于基因组(WGS+MIRU-VNTR+基本病例数据)的调查,以确定每个人 TB 的最可能来源,并使用在线、多项选择调查(n=4)和现场组访谈(n=5)评估方案人员对基因分型和基因组学的知识、态度和做法。现场和基于基因组的方法在 32 例(81%)可能发生结核病的情况下达成了 26 例(81%)的协议。在确定具体的源病例方面,一致性较差(13/22 或 59%的病例)。单一位点 MIRU-VNTR 变体和有限的遗传多样性使分析变得复杂。定性数据表明,参与者认为基因组流行病学是一种有用的工具,可以简化调查,特别是在区分潜伏性 TB 再激活与近期传播方面。基于此,基因组数据可用于增强 CI、集中资源、靶向干预措施和帮助结核病规划评估。