Crop Improvement Division, ICAR-National Rice Research Institute (Formerly CRRI), Cuttack-753006, Odisha, India.
Int J Mol Sci. 2020 Feb 6;21(3):1080. doi: 10.3390/ijms21031080.
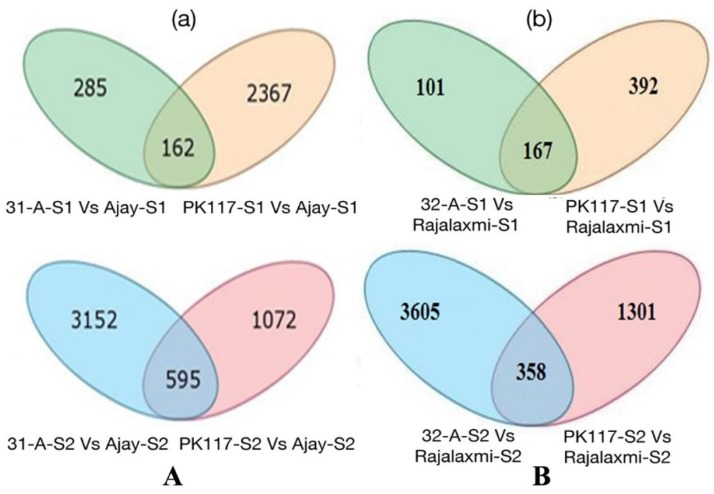
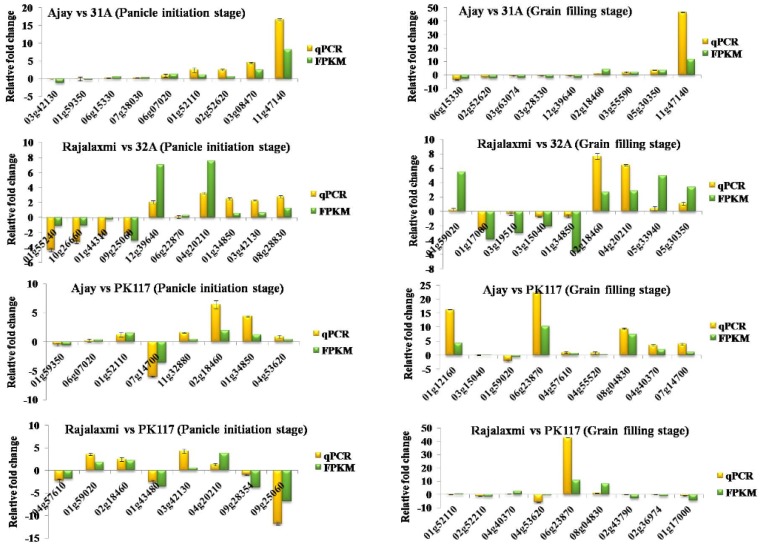
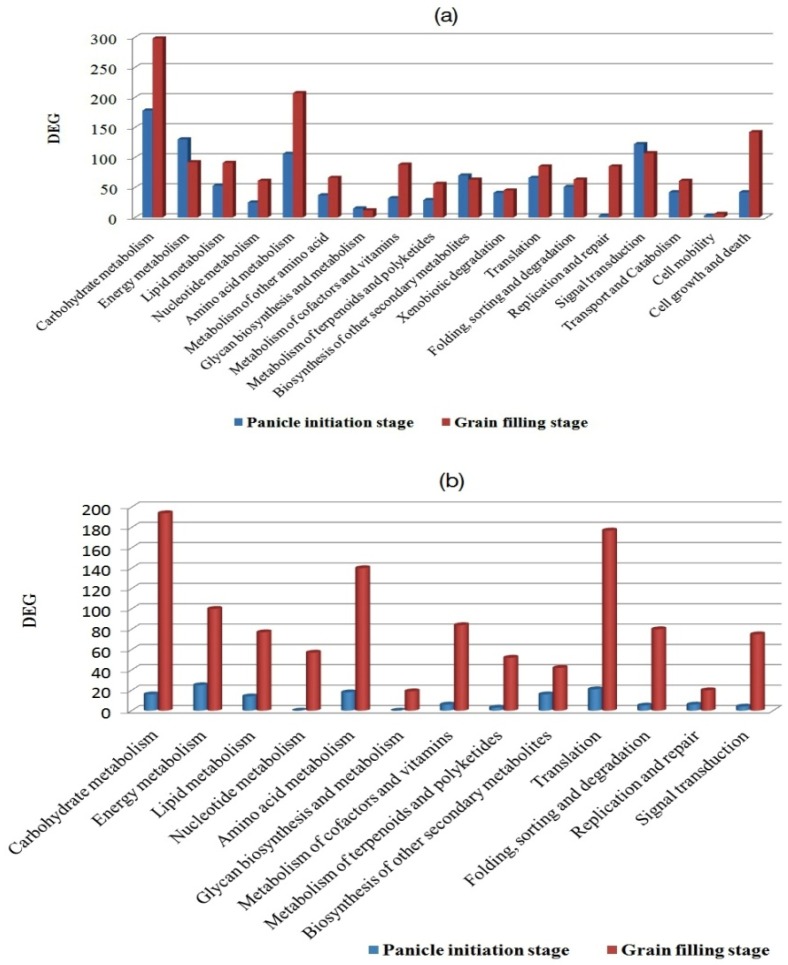
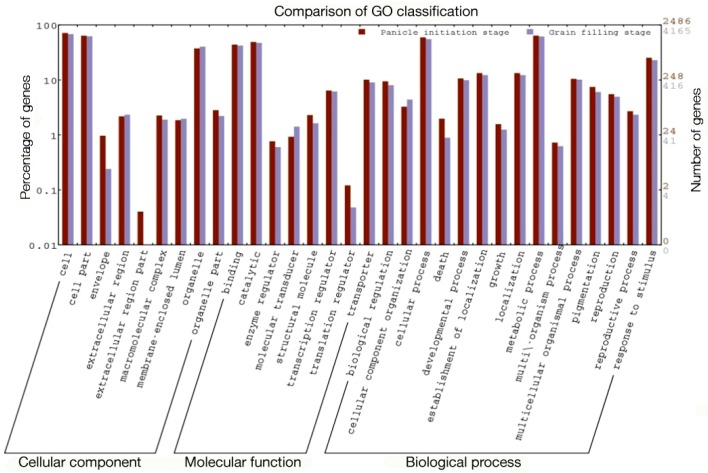
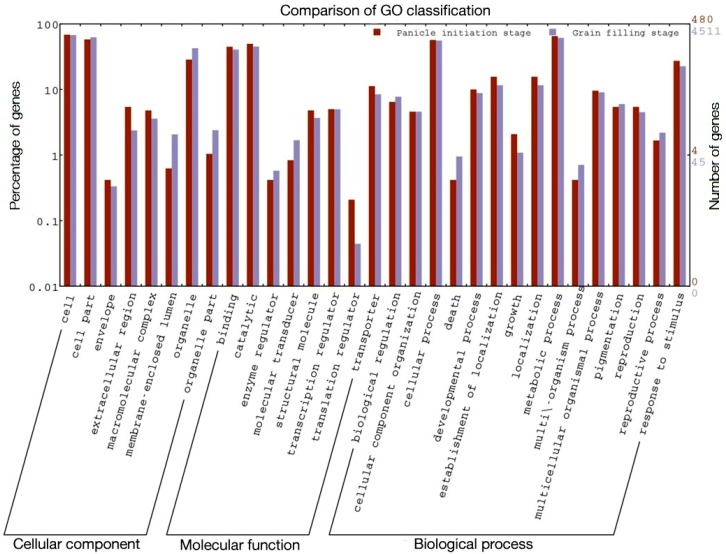
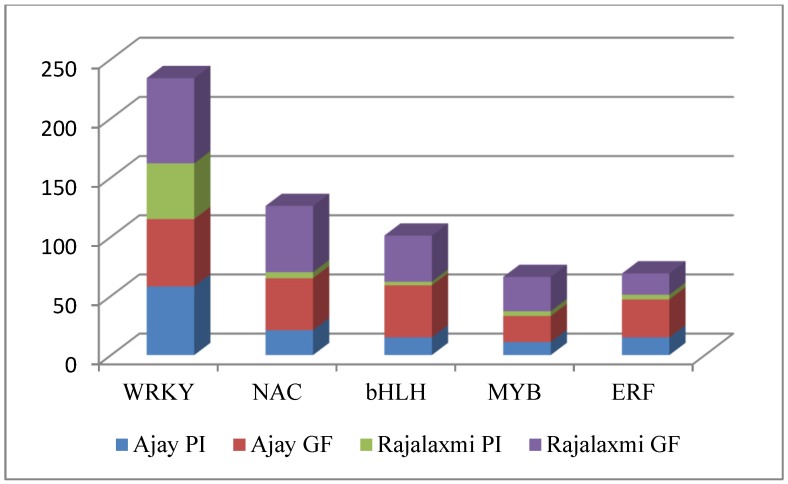
RNA-Seq technology was used to analyze the transcriptome of two rice hybrids, Ajay (based on wild-abortive (WA)-cytoplasm) and Rajalaxmi (based on Kalinga-cytoplasm), and their respective parents at the panicle initiation (PI) and grain filling (GF) stages. Around 293 and 302 million high quality paired-end reads of Ajay and Rajalaxmi, respectively, were generated and aligned against the Nipponbare reference genome. Transcriptome profiling of Ajay revealed 2814 and 4819 differentially expressed genes (DEGs) at the PI and GF stages, respectively, as compared to its parents. In the case of Rajalaxmi, 660 and 5264 DEGs were identified at PI and GF stages, respectively. Functionally relevant DEGs were selected for validation through qRT-PCR, which were found to be co-related with the expression patterns to RNA-seq. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis indicated significant DEGs enriched for energy metabolism pathways, such as photosynthesis, oxidative phosphorylation, and carbon fixation, at the PI stage, while carbohydrate metabolism-related pathways, such as glycolysis and starch and sucrose metabolism, were significantly involved at the GF stage. Many genes involved in energy metabolism exhibited upregulation at the PI stage, whereas the genes involved in carbohydrate biosynthesis had higher expression at the GF stage. The majority of the DEGs were successfully mapped to know yield related rice quantitative trait loci (QTLs). A set of important transcription factors (TFs) was found to be encoded by the identified DEGs. Our results indicated that a complex interplay of several genes in different pathways contributes to higher yield and vigor in rice hybrids.
利用 RNA-Seq 技术分析了两个水稻杂种(基于野生败育(WA)细胞质的 Ajay 和基于 Kalinga 细胞质的 Rajalaxmi)及其亲本在小穗起始(PI)和灌浆(GF)阶段的转录组。分别为 Ajay 和 Rajalaxmi 生成了约 2.93 亿和 3.02 亿对高质量的配对末端reads,并与 Nipponbare 参考基因组进行了比对。与亲本相比,Ajay 在 PI 和 GF 阶段分别有 2814 个和 4819 个差异表达基因(DEGs)。在 Rajalaxmi 中,在 PI 和 GF 阶段分别鉴定出 660 个和 5264 个 DEGs。通过 qRT-PCR 对功能相关的 DEGs 进行了验证,结果发现这些基因的表达模式与 RNA-seq 一致。京都基因与基因组百科全书(KEGG)通路分析表明,在 PI 阶段,与能量代谢途径相关的显著 DEGs 富集,如光合作用、氧化磷酸化和碳固定,而在 GF 阶段,与碳水化合物代谢相关的途径,如糖酵解和淀粉和蔗糖代谢,也显著参与。许多参与能量代谢的基因在 PI 阶段上调,而参与碳水化合物生物合成的基因在 GF 阶段表达更高。大多数 DEGs 成功映射到已知的与水稻产量相关的数量性状位点(QTLs)。发现一组由鉴定的 DEGs 编码的重要转录因子(TFs)。我们的研究结果表明,不同途径的多个基因之间的复杂相互作用有助于提高水稻杂种的产量和活力。