Ibrahim Muhammad Tukur, Uzairu Adamu, Shallangwa Gideon Adamu, Uba Sani
Department of Chemistry, Faculty of Physical Science, Ahmadu Bello University, P.M.B 1045, Zaria, Kaduna State Nigeria.
Heliyon. 2020 Jan 7;6(1):e03158. doi: 10.1016/j.heliyon.2020.e03158. eCollection 2020 Jan.
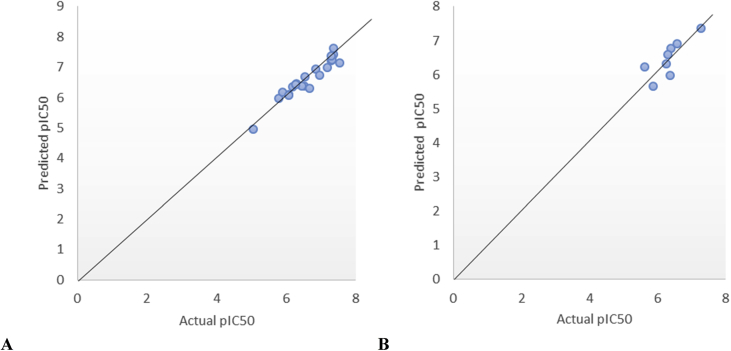
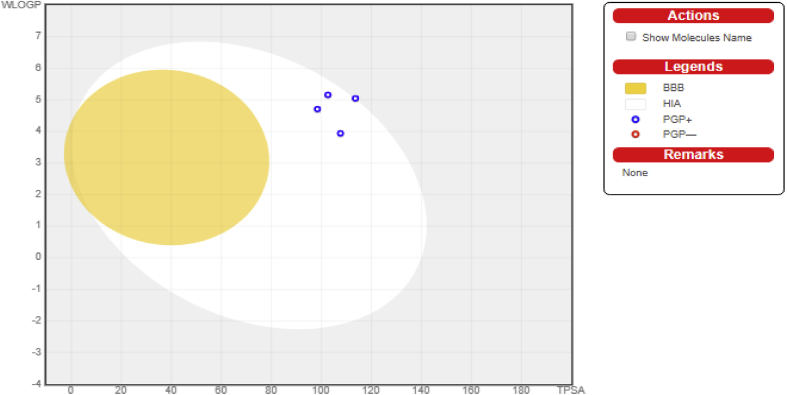
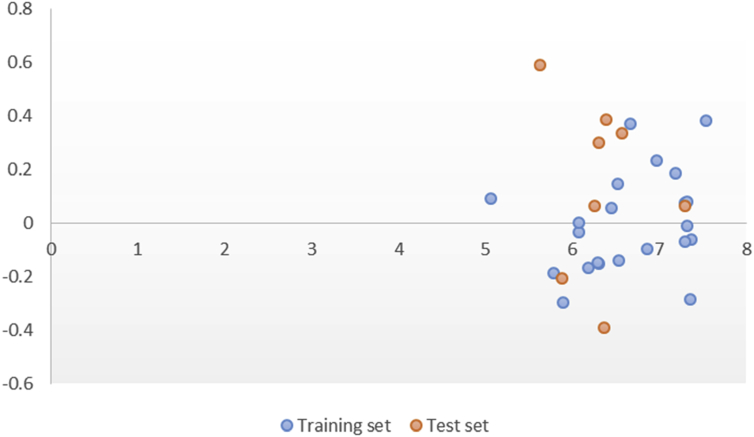
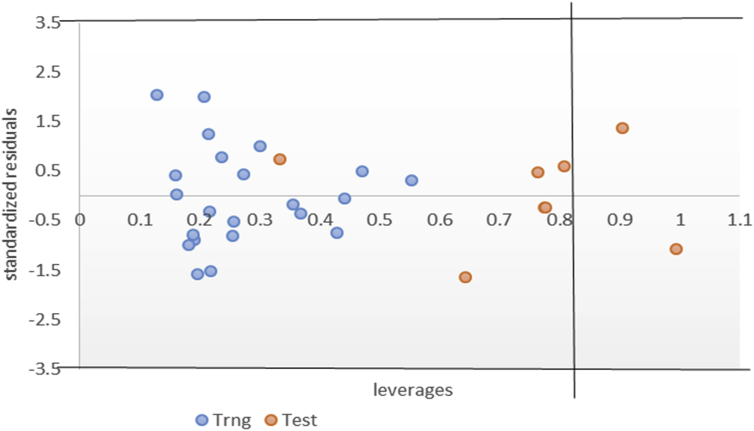
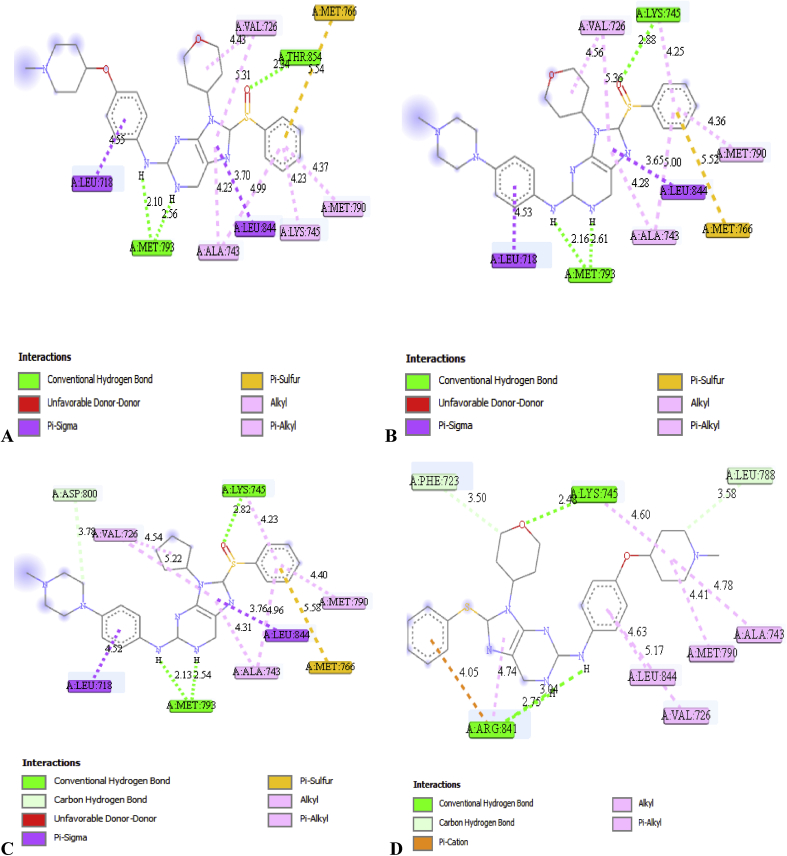
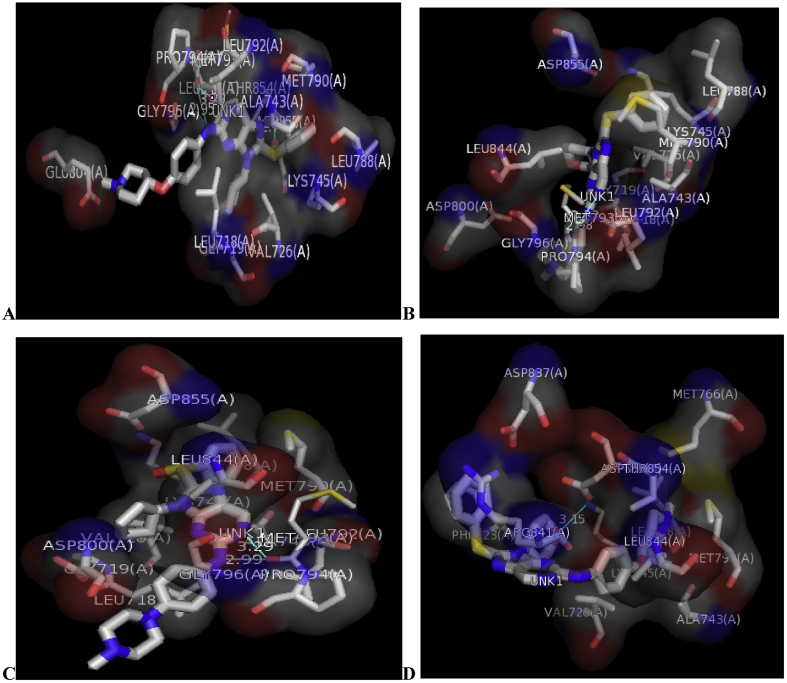
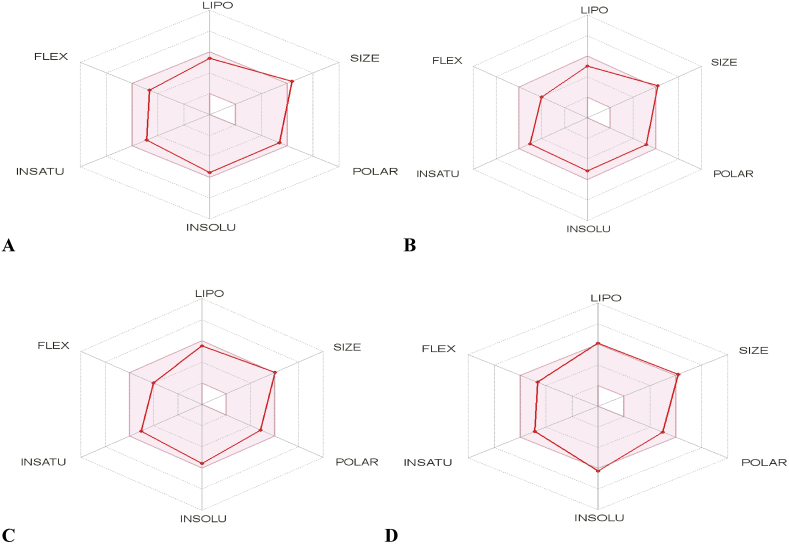
In-silico activity prediction was performed to predict new inhibitory activities of 2, 9-disubstituted 8-phenylthio/phenylsulfinyl-9h-purine derivatives as anti-proliferative agents using QSAR technique. The anti-proliferative agents were optimized using Density Functional Theory (DFT) method utilizing the B3LYP/6-31G* level of theory. Genetic Function Algorithm (GFA) was used to build the QSAR models. Out of the models built, the best one was selected and reported because of its fitness statistically with the following assessment parameters: R = 0.919035, R = 0.893733, Q = 0.866475, R = 0.636217, and LOF = 0.215884. The selected model was further subjected to other assessment such as VIF, Y-scrambling test, applicability domain and found to be statistically significant. The binding mode of some selected 2, 9-disubstituted 8-phenylthio/phenylsulfinyl-9H-purine (ligands) in the active site of EGFR-tyrosine kinase (EGFR-TK) (receptor) was studied via Molecular docking. Molecule 22 was identified to have the highest binding energy (-10.4 kcal/mol) among the other selected ligands which it might be as a result of hydrogen interactions formed with MET793 (2.48599 Å, 2.04522 Å) & THR854 (3.76616 Å) amino acid residues and hydrophobic/other interactions with amino acid residues (LEU718, LEU844, MET766, VAL726, ALA743, LYS745 and MET790) in the active site of EGFR-tyrosine kinase (EGFR-TK). The drug-likeness of these selected anti-proliferative agents were predicted via the pharmacokinetics profile of the molecules utilizing SWISS ADME. The anti-proliferative agents were found to be orally safe by not having more than 1 violation of the Lipinski's rule of five. This research proposed a way for designing potent anti-proliferative agents against their target enzyme.
利用定量构效关系(QSAR)技术进行了计算机模拟活性预测,以预测2,9-二取代的8-苯硫基/苯亚砜基-9H-嘌呤衍生物作为抗增殖剂的新抑制活性。使用密度泛函理论(DFT)方法,在B3LYP/6-31G*理论水平下对抗增殖剂进行了优化。采用遗传函数算法(GFA)建立QSAR模型。在所建立的模型中,选择并报告了最佳模型,因为其在统计学上与以下评估参数拟合良好:R = 0.919035,R = 0.893733,Q = 0.866475,R = 0.636217,以及LOF = 0.215884。所选模型进一步接受了其他评估,如方差膨胀因子(VIF)、Y-随机化检验、适用域,结果发现具有统计学意义。通过分子对接研究了一些选定的2,9-二取代的8-苯硫基/苯亚砜基-9H-嘌呤(配体)在表皮生长因子受体酪氨酸激酶(EGFR-TK)(受体)活性位点的结合模式。分子22在其他选定配体中被确定具有最高的结合能(-10.4千卡/摩尔),这可能是由于它与MET793(2.48599 Å,2.04522 Å)和THR854(3.76616 Å)氨基酸残基形成了氢键相互作用,以及与表皮生长因子受体酪氨酸激酶(EGFR-TK)活性位点的氨基酸残基(LEU718、LEU844、MET766、VAL726、ALA743、LYS745和MET790)形成了疏水/其他相互作用。利用SWISS ADME通过分子的药代动力学特征预测了这些选定抗增殖剂的类药性。这些抗增殖剂被发现口服安全,因为它们违反Lipinski五规则的情况不超过1次。本研究提出了一种针对其靶酶设计强效抗增殖剂的方法。