Department of Bio-Engineering, School of Engineering, Vels Institute of Science Technology and Advanced Studies, Chennai, Tamil Nadu, India.
Department of Biotechnology, Rajalakshmi Engineering College, Thandalam, Tamil Nadu, India.
J Biomol Struct Dyn. 2022 Feb;40(2):585-611. doi: 10.1080/07391102.2020.1815584. Epub 2020 Sep 8.
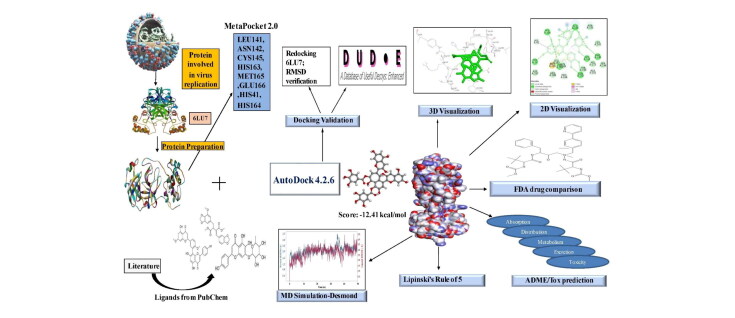
The study aims to evaluate the potency of two hundred natural antiviral phytocompounds against the active site of the Severe Acquired Respiratory Syndrome - Coronavirus - 2 (SARS-CoV-2) Main-Protease (M) using AutoDock 4.2.6. The three- dimensional crystal structure of the M (PDB Id: 6LU7) was retrieved from the Protein Data Bank (PDB), the active site was predicted using MetaPocket 2.0. Food and Drug Administration (FDA) approved viral protease inhibitors were used as standards for comparison of results. The compounds theaflavin-3-3'-digallate, rutin, hypericin, robustaflavone, and (-)-solenolide A with respective binding energy of -12.41 (Ki = 794.96 pM); -11.33 (Ki = 4.98 nM); -11.17 (Ki = 6.54 nM); -10.92 (Ki = 9.85 nM); and -10.82 kcal/mol (Ki = 11.88 nM) were ranked top as Coronavirus Disease - 2019 (COVID-19) M inhibitors. The interacting amino acid residues were visualized using Discovery Studio 3.5 to elucidate the 2-dimensional and 3-dimensional interactions. The study was validated by i) re-docking the N3-peptide inhibitor-M and superimposing them onto co-crystallized complex and ii) docking decoy ligands to M. The ligands that showed low binding energy were further predicted for and pharmacokinetic properties and Lipinski's rule of 5 and the results are tabulated and discussed. Molecular dynamics simulations were performed for 50 ns for those compounds using the Desmond package, Schrödinger to assess the conformational stability and fluctuations of protein-ligand complexes during the simulation. Thus, the natural compounds could act as a lead for the COVID-19 regimen after and clinical trials.Communicated by Ramaswamy H. Sarma.
本研究旨在使用 AutoDock 4.2.6 评估 200 种天然抗病毒植物化合物对严重急性呼吸系统综合征 - 冠状病毒 2 (SARS-CoV-2) 主要蛋白酶 (M) 的活性部位的效力。M 的三维晶体结构 (PDB ID: 6LU7) 从蛋白质数据银行 (PDB) 中检索出来,使用 MetaPocket 2.0 预测活性部位。食品和药物管理局 (FDA) 批准的病毒蛋白酶抑制剂被用作比较结果的标准。化合物表没食子儿茶素-3-3'-二没食子酸酯、芦丁、金丝桃素、罗伯斯特黄酮和 (-)-索勒诺内酯 A 的结合能分别为-12.41 (Ki = 794.96 pM);-11.33 (Ki = 4.98 nM);-11.17 (Ki = 6.54 nM);-10.92 (Ki = 9.85 nM);和-10.82 kcal/mol (Ki = 11.88 nM),被列为冠状病毒病 2019 (COVID-19) M 抑制剂的前 5 名。使用 Discovery Studio 3.5 可视化相互作用的氨基酸残基,以阐明 2 维和 3 维相互作用。该研究通过以下方法进行验证:i) 重新对接 N3-肽抑制剂-M,并将其与共结晶复合物叠加,ii) 将诱饵配体对接至 M。对显示低结合能的配体进行进一步预测,并对其进行药代动力学特性和 Lipinski 的 5 规则分析,结果列于表中并进行讨论。使用 Desmond 包和 Schrödinger 对这些化合物进行了 50 ns 的分子动力学模拟,以评估蛋白质-配体复合物在模拟过程中的构象稳定性和波动。因此,天然化合物在和临床试验后可能成为 COVID-19 治疗方案的先导。由 Ramaswamy H. Sarma 传达。