Clinical Research Department, London School of Hygiene and Tropical Medicine, London, UK.
Microbiology Department, Great Ormond Street Hospital NHS Foundation Trust, London, UK.
Microb Genom. 2020 Feb;6(2). doi: 10.1099/mgen.0.000335. Epub 2020 Feb 12.
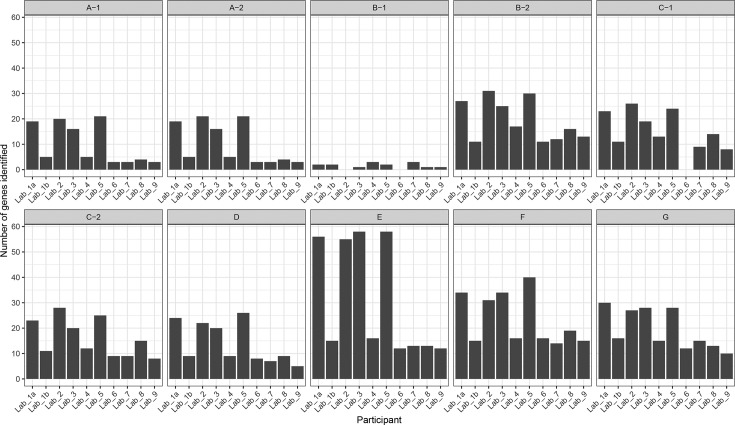
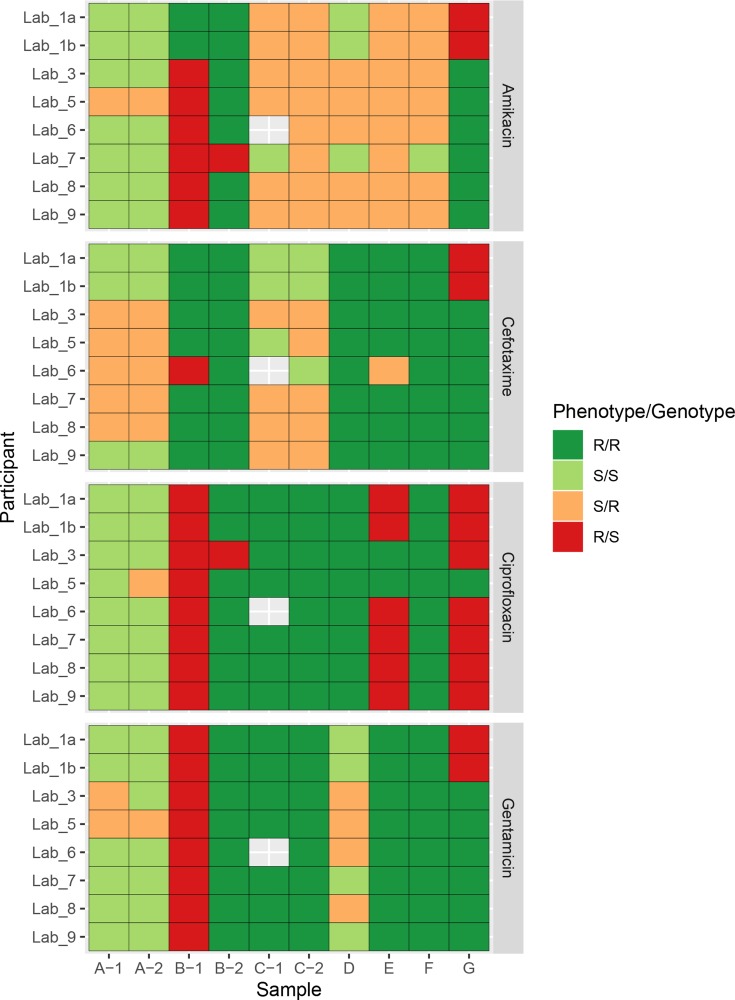
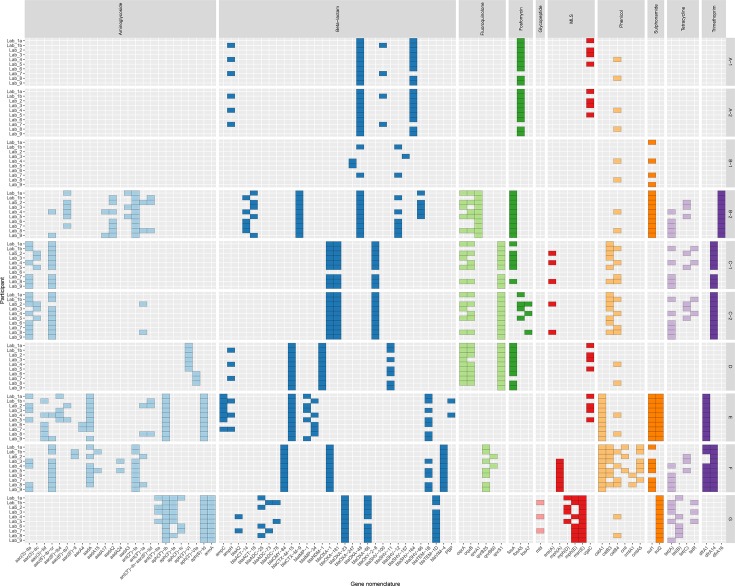
Antimicrobial resistance (AMR) poses a threat to public health. Clinical microbiology laboratories typically rely on culturing bacteria for antimicrobial-susceptibility testing (AST). As the implementation costs and technical barriers fall, whole-genome sequencing (WGS) has emerged as a 'one-stop' test for epidemiological and predictive AST results. Few published comparisons exist for the myriad analytical pipelines used for predicting AMR. To address this, we performed an inter-laboratory study providing sets of participating researchers with identical short-read WGS data from clinical isolates, allowing us to assess the reproducibility of the bioinformatic prediction of AMR between participants, and identify problem cases and factors that lead to discordant results. We produced ten WGS datasets of varying quality from cultured carbapenem-resistant organisms obtained from clinical samples sequenced on either an Illumina NextSeq or HiSeq instrument. Nine participating teams ('participants') were provided these sequence data without any other contextual information. Each participant used their choice of pipeline to determine the species, the presence of resistance-associated genes, and to predict susceptibility or resistance to amikacin, gentamicin, ciprofloxacin and cefotaxime. We found participants predicted different numbers of AMR-associated genes and different gene variants from the same clinical samples. The quality of the sequence data, choice of bioinformatic pipeline and interpretation of the results all contributed to discordance between participants. Although much of the inaccurate gene variant annotation did not affect genotypic resistance predictions, we observed low specificity when compared to phenotypic AST results, but this improved in samples with higher read depths. Had the results been used to predict AST and guide treatment, a different antibiotic would have been recommended for each isolate by at least one participant. These challenges, at the final analytical stage of using WGS to predict AMR, suggest the need for refinements when using this technology in clinical settings. Comprehensive public resistance sequence databases, full recommendations on sequence data quality and standardization in the comparisons between genotype and resistance phenotypes will all play a fundamental role in the successful implementation of AST prediction using WGS in clinical microbiology laboratories.
抗菌药物耐药性(AMR)对公众健康构成威胁。临床微生物学实验室通常依赖于培养细菌进行抗菌药物敏感性测试(AST)。随着实施成本和技术障碍的降低,全基因组测序(WGS)已成为一种用于进行流行病学和预测性 AST 结果的“一站式”测试。目前,针对用于预测 AMR 的众多分析管道,发表的比较研究还很少。为了解决这个问题,我们进行了一项实验室间研究,为参与研究的人员提供了来自临床分离物的相同短读 WGS 数据,使我们能够评估参与者之间 AMR 生物信息预测的可重复性,并确定导致结果不一致的问题案例和因素。我们从临床样本中培养出的碳青霉烯类耐药菌中产生了十个质量不同的 WGS 数据集,这些样本是在 Illumina NextSeq 或 HiSeq 仪器上测序的。九个参与团队(“参与者”)获得了这些没有任何其他上下文信息的序列数据。每个参与者都使用自己选择的管道来确定物种、存在耐药相关基因,并预测对阿米卡星、庆大霉素、环丙沙星和头孢噻肟的敏感性或耐药性。我们发现,参与者从相同的临床样本中预测了不同数量的 AMR 相关基因和不同的基因变体。序列数据的质量、生物信息学管道的选择以及结果的解释都导致了参与者之间的不一致。尽管大部分不准确的基因变体注释不会影响基因型耐药预测,但与表型 AST 结果相比,我们观察到特异性较低,但在读取深度较高的样本中有所提高。如果将这些结果用于预测 AST 并指导治疗,那么至少有一位参与者会针对每个分离物推荐不同的抗生素。在使用 WGS 预测 AMR 的最终分析阶段,这些挑战表明在临床环境中使用该技术时需要进行改进。全面的公共耐药序列数据库、关于序列数据质量的全面建议以及基因型与耐药表型之间的比较标准化都将在临床微生物学实验室中成功实施 WGS 预测 AST 方面发挥重要作用。