Jiang Yao, Tang Shaoqing, Xiao Wei, Yun Peng, Ding Xiangdong
National Engineering Laboratory for Animal Breeding, Laboratory of Animal Genetics, Breeding and Reproduction, Ministry of Agriculture, College of Animal Science and Technology, China Agricultural University, Beijing 100193, China.
Beijing Station of Animal Husbandry, Beijing 100107, China.
Asian-Australas J Anim Sci. 2020 Sep;33(9):1400-1410. doi: 10.5713/ajas.19.0411. Epub 2019 Oct 21.
Genome-wide association study and two meta-analysis based on GWAS performed to explore the genetic mechanism underlying variation in pig number born alive (NBA) and total number born (TNB).
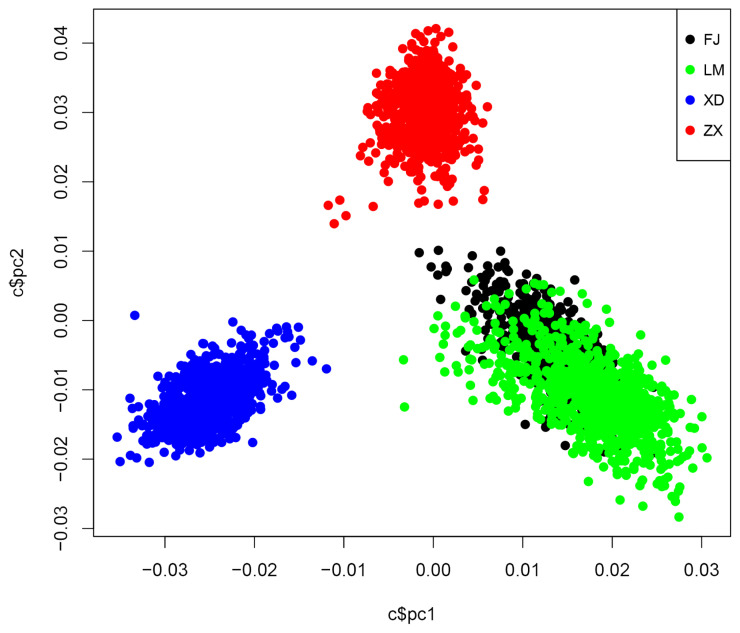
Single trait GWAS and two meta-analysis (single-trait meta analysis and multitrait meta analysis) were used in our study for NBA and TNB on 3,121 Yorkshires from 4 populations, including three different American Yorkshire populations (n = 2,247) and one British Yorkshire populations (n = 874).
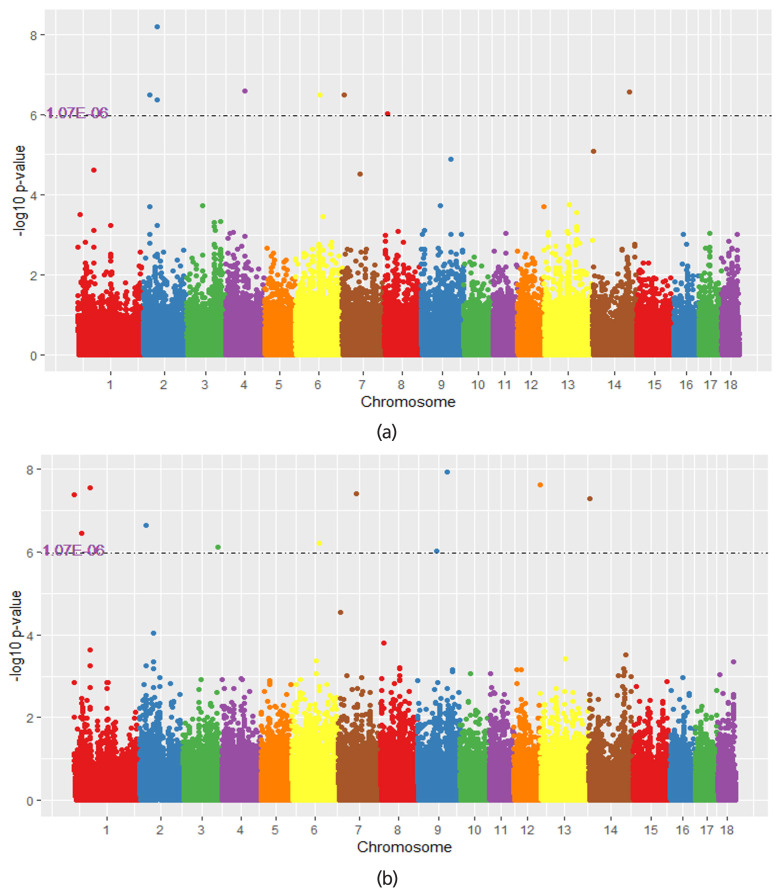
The result of single trait GWAS showed that no significant associated single nucleotide polymorphisms (SNPs) were identified. Using single-trait meta analysis and multi-trait meta analysis within populations, 11 significant loci were identified associated with target traits. Spindlin 1, vascular endothelial growth factor A, forkhead box Q1, msh homeobox 1, and LHFPL tetraspan submily member 3 are five functionally plausible candidate genes for NBA and TNB. Compared to the single population GWAS, single-trait Meta analysis can improve the detection power to identify SNPs by integrating information of multiple populations. The multiple-trait analysis reduced the power to detect trait-specific loci but enhanced the power to identify the common loci across traits.
In total, our findings identified novel genes to be validated as candidates for NBA and TNB in pigs. Also, it enabled us to enlarge population size by including multiple populations with different genetic backgrounds and increase the power of GWAS by using meta analysis.
开展全基因组关联研究以及基于全基因组关联研究的两项荟萃分析,以探究产活仔数(NBA)和总产仔数(TNB)变异背后的遗传机制。
本研究对来自4个群体的3121头约克夏猪的NBA和TNB进行了单性状全基因组关联研究以及两项荟萃分析(单性状荟萃分析和多性状荟萃分析),这4个群体包括三个不同的美国约克夏群体(n = 2247)和一个英国约克夏群体(n = 874)。
单性状全基因组关联研究结果显示,未鉴定出显著相关的单核苷酸多态性(SNP)。在群体内部使用单性状荟萃分析和多性状荟萃分析,鉴定出11个与目标性状相关的显著位点。纺锤体相关蛋白1、血管内皮生长因子A、叉头框Q1、msh同源盒1和LHFPL四跨膜蛋白亚家族成员3是NBA和TNB的五个功能上合理的候选基因。与单群体全基因组关联研究相比,单性状荟萃分析通过整合多个群体的信息可以提高识别SNP的检测能力。多性状分析降低了检测性状特异性位点的能力,但增强了识别跨性状共同位点的能力。
总的来说,我们的研究结果鉴定出了可作为猪NBA和TNB候选基因进行验证的新基因。此外,通过纳入具有不同遗传背景的多个群体,我们扩大了群体规模,并通过荟萃分析提高了全基因组关联研究的效力。