Antimicrobial Resistance and Infectious Diseases Research Laboratory, Murdoch University, Murdoch, Australia.
Statens Serum Institute, Copenhagen, Denmark.
PLoS One. 2020 Feb 14;15(2):e0228781. doi: 10.1371/journal.pone.0228781. eCollection 2020.
Over the last three decades, hospital adapted clonal complex (CC) 17 strains of Enterococcus faecium have acquired and exchanged antimicrobial resistance genes leading to the widespread resistance to clinically important antimicrobials globally. In Australia, a high prevalence of vancomycin resistance has been reported in E. faecium in the last decade.
In this study, we determined the phylogenetic relationship and genetic characteristics of E. faecium collected from hospitalized patients with blood stream infections throughout Australia from 2015 to 2017 using high throughput molecular techniques.
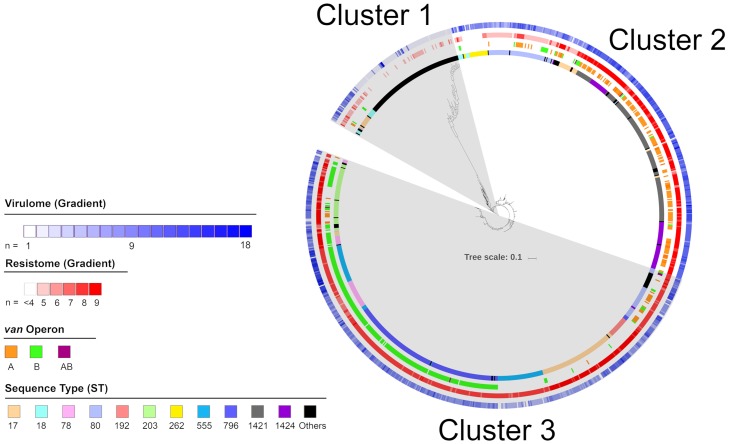
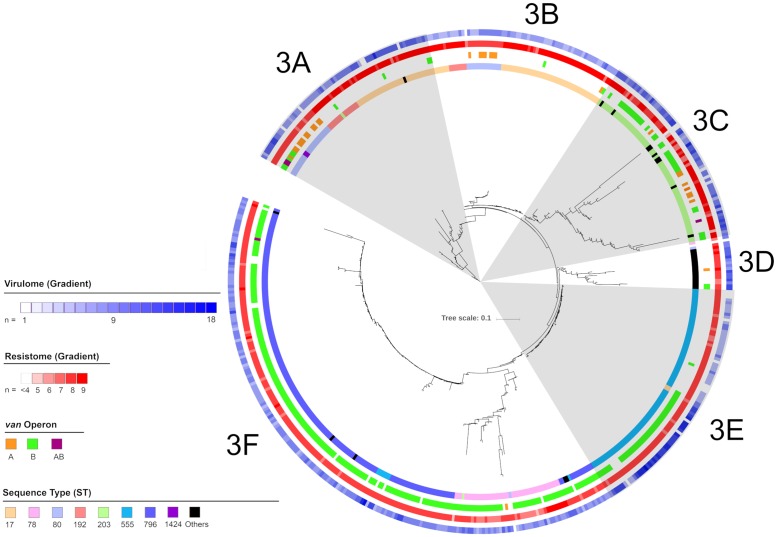
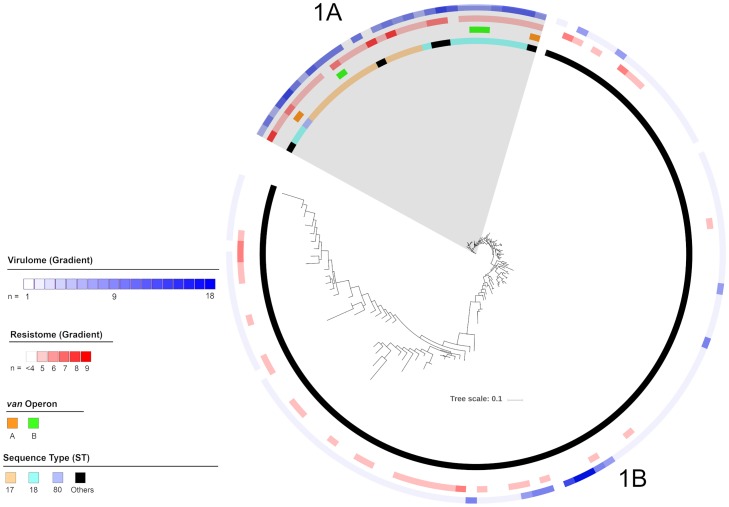
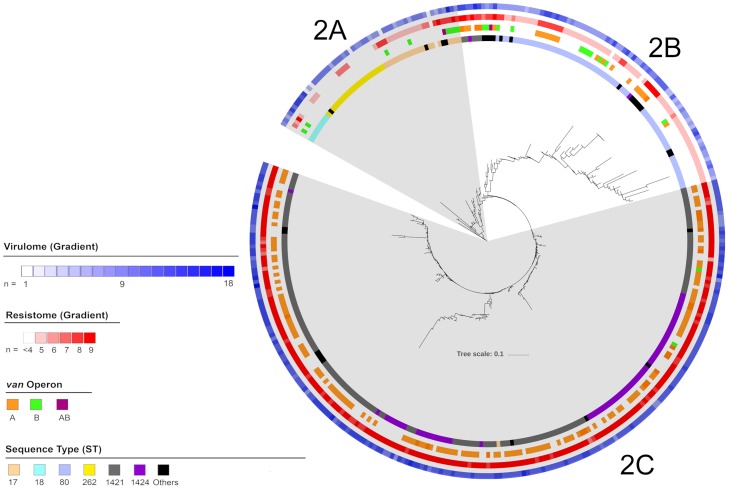
Using single nucleotide polymorphism based phylogenetic inference, three distinct clusters of isolates were observed with additional sub-clustering. One cluster harboured mostly non-CC17 isolates while two clusters were dominant for the vanA and vanB operons.
The gradual increase in dominance of the respective van operon was observed in both the vanA and vanB dominant clusters suggesting a strain-van operon affinity. The high prevalence of the van operon within isolates of a particular sub-cluster was linked to an increased number of isolates and 30-day all-cause mortality. Different dominant sub-clusters were observed in each region of Australia. Findings from this study can be used to put future surveillance data into a broader perspective including the detection of novel E. faecium strains in Australia as well as the dissemination and evolution of each strain.
在过去的三十年中,医院适应的粪肠球菌克隆复合体(CC)17 株已获得并交换了导致全球广泛对抗菌药物产生耐药性的抗生素耐药基因。在澳大利亚,过去十年中报告了粪肠球菌对万古霉素的高耐药率。
本研究采用高通量分子技术,对 2015 年至 2017 年期间澳大利亚住院患者血流感染中分离的粪肠球菌进行了遗传特征和系统发育关系的研究。
通过基于单核苷酸多态性的系统发育推断,观察到三个不同的分离株簇,还有额外的亚聚类。一个簇主要包含非 CC17 分离株,而两个簇主要包含 vanA 和 vanB 操纵子。
vanA 和 vanB 优势簇中各自 van 操纵子的优势逐渐增加,表明存在菌株-van 操纵子亲和力。特定亚簇内的 van 操纵子的高流行率与增加的分离株数量和 30 天全因死亡率有关。在澳大利亚的每个地区都观察到不同的优势亚簇。本研究的结果可以用于将未来的监测数据置于更广泛的背景下,包括在澳大利亚检测新型粪肠球菌菌株以及每个菌株的传播和进化。