Medical Research Council Cancer Unit, University of Cambridge, Hills Road, Cambridge CB2 0XZ, UK.
Medical Research Council Cancer Unit, University of Cambridge, Hills Road, Cambridge CB2 0XZ, UK.
Cell Rep. 2020 Feb 18;30(7):2083-2093.e5. doi: 10.1016/j.celrep.2020.01.074.
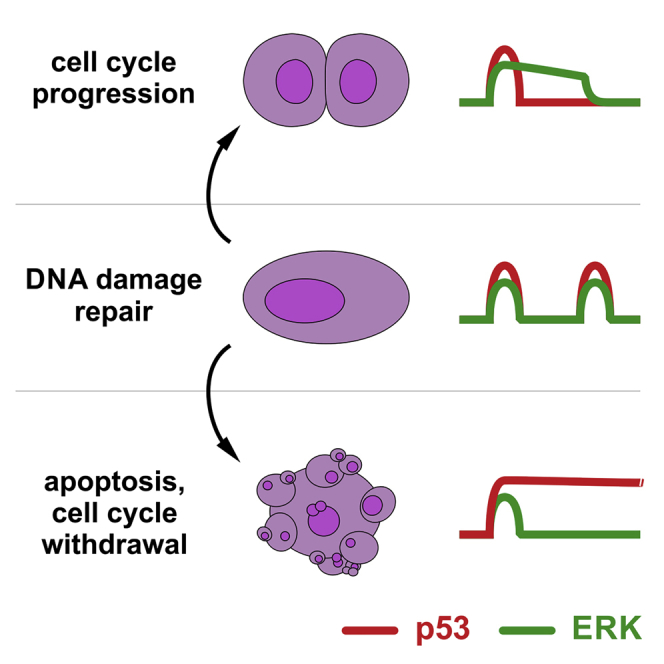
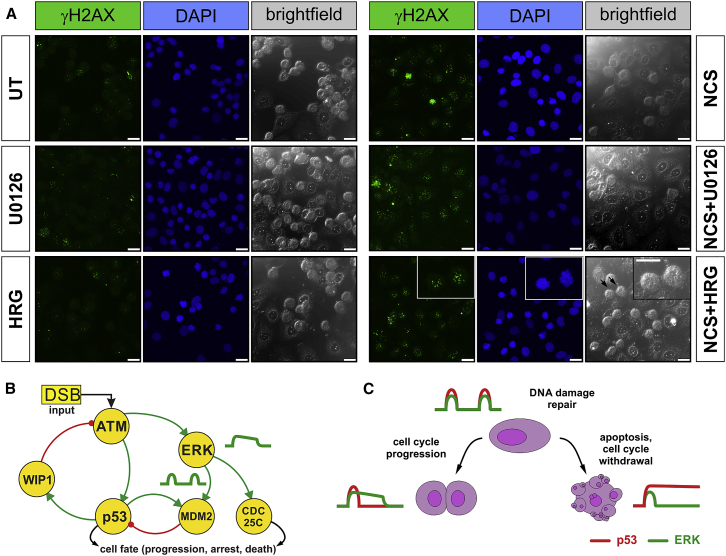
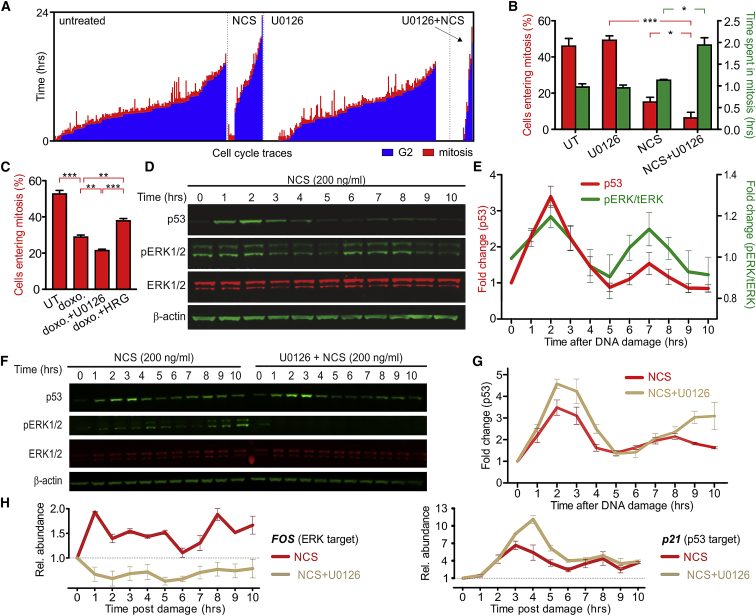
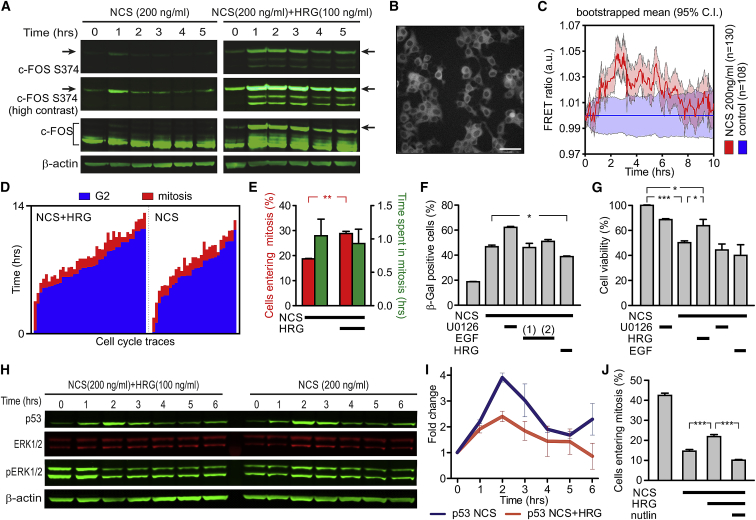
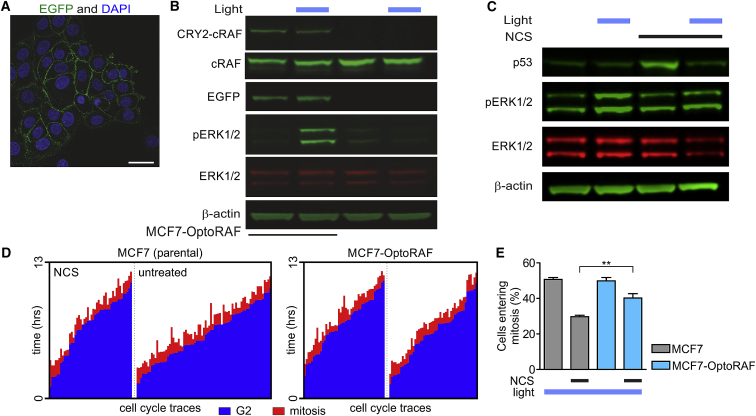
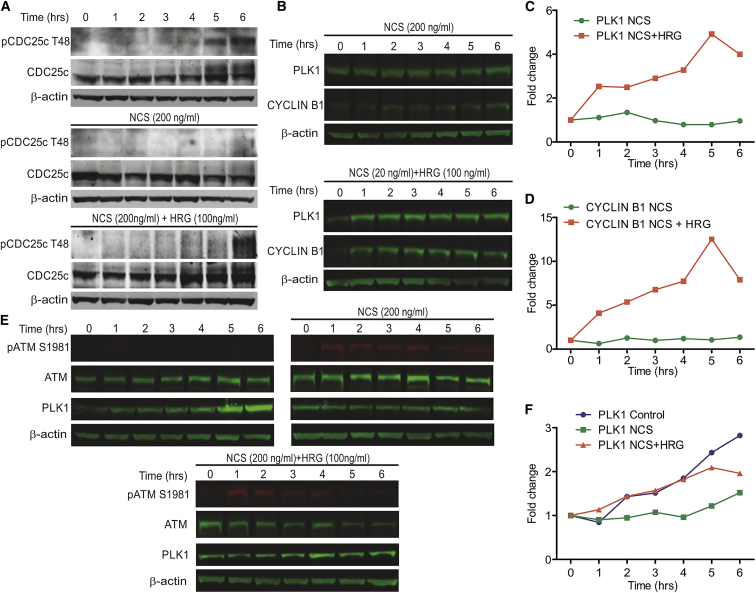
Cell-autonomous changes in p53 expression govern the duration and outcome of cell-cycle arrest at the G2 checkpoint for DNA damage. Here, we report that mitogen-activated protein kinase (MAPK) signaling integrates extracellular cues with p53 dynamics to determine cell fate at the G2 checkpoint. Optogenetic tools and quantitative cell biochemistry reveal transient oscillations in MAPK activity dependent on ataxia-telangiectasia-mutated kinase after DNA damage. MAPK inhibition alters p53 dynamics and p53-dependent gene expression after checkpoint enforcement, prolonging G2 arrest. In contrast, sustained MAPK signaling induces the phosphorylation of CDC25C, and consequently, the accumulation of pro-mitotic kinases, thereby relaxing checkpoint stringency and permitting cells to evade prolonged G2 arrest and senescence induction. We propose a model in which this MAPK-mediated mechanism integrates extracellular cues with cell-autonomous p53-mediated signals, to safeguard genomic integrity during tissue proliferation. Early steps in oncogene-driven carcinogenesis may imbalance this tumor-suppressive mechanism to trigger genome instability.
细胞自主的 p53 表达变化控制着 DNA 损伤时 G2 检查点细胞周期阻滞的持续时间和结果。在这里,我们报告称,有丝分裂原激活的蛋白激酶(MAPK)信号转导将细胞外信号与 p53 动力学整合在一起,以确定 G2 检查点的细胞命运。光遗传学工具和定量细胞生物化学揭示了 DNA 损伤后依赖共济失调毛细血管扩张突变激酶(ataxia-telangiectasia-mutated kinase,ATM)的 MAPK 活性的短暂振荡。MAPK 抑制改变了检查点执行后 p53 动力学和 p53 依赖性基因表达,延长了 G2 期阻滞。相比之下,持续的 MAPK 信号诱导 CDC25C 的磷酸化,进而导致促有丝分裂激酶的积累,从而放松检查点的严格性,允许细胞逃避长时间的 G2 期阻滞和衰老诱导。我们提出了一个模型,其中这个 MAPK 介导的机制将细胞外信号与细胞自主的 p53 介导的信号整合在一起,以在组织增殖过程中保护基因组的完整性。癌基因驱动的致癌作用的早期步骤可能会使这种肿瘤抑制机制失去平衡,从而引发基因组不稳定性。