Khaledian Ehdieh, Brayton Kelly A, Broschat Shira L
School of Electrical Engineering and Computer Science, Washington State University, P.O. Box 642752, Pullman, WA 99164, USA.
Department of Veterinary Microbiology and Pathology, Washington State University, P.O. Box 647040, Pullman, WA 99164, USA.
Microorganisms. 2020 Feb 24;8(2):312. doi: 10.3390/microorganisms8020312.
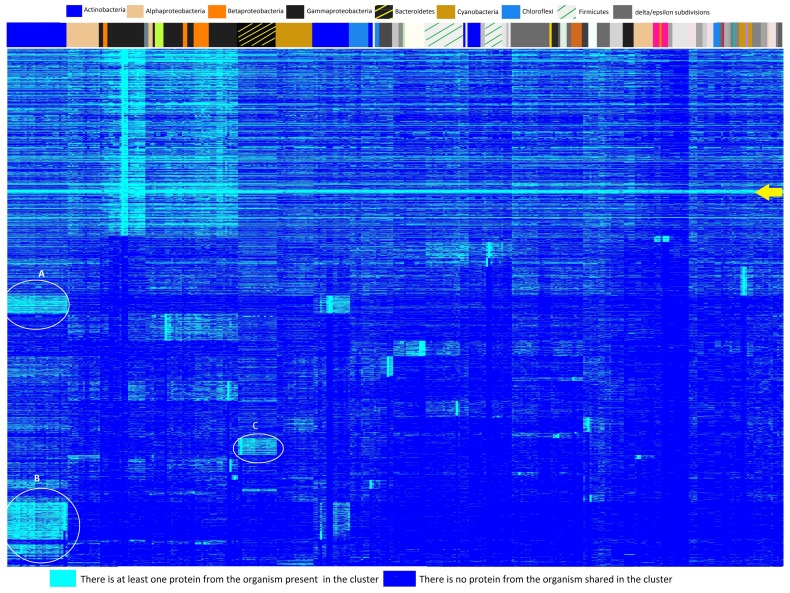
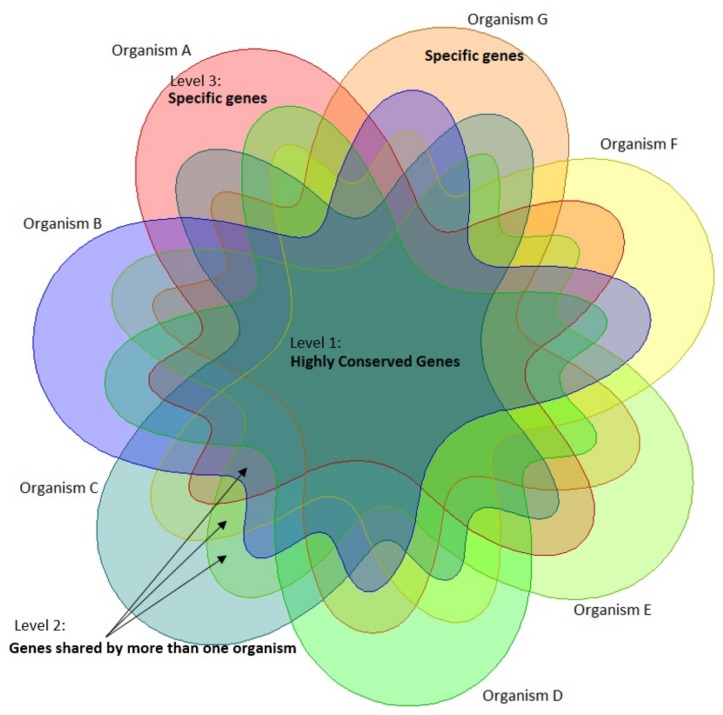
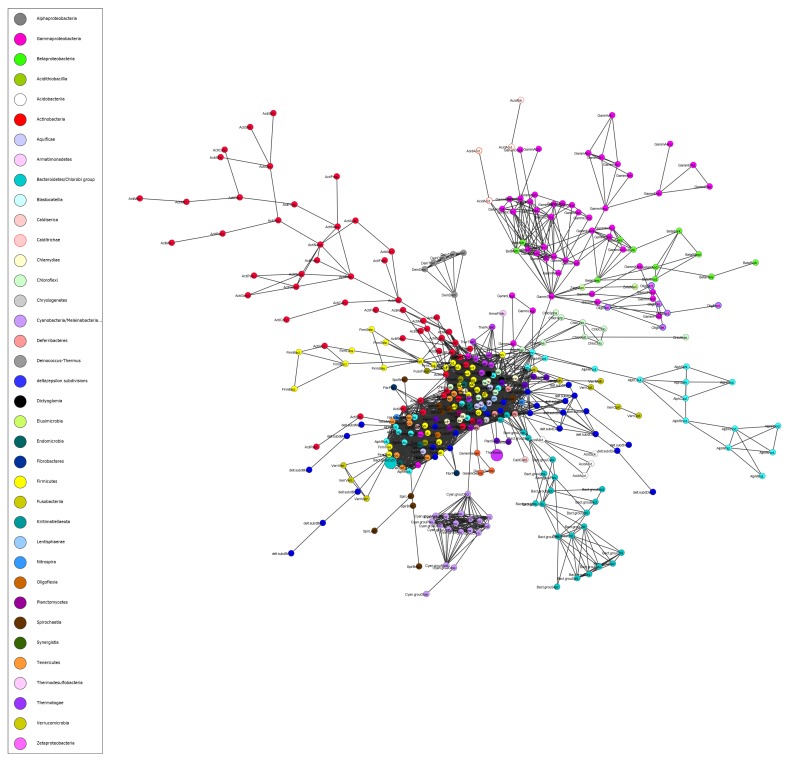
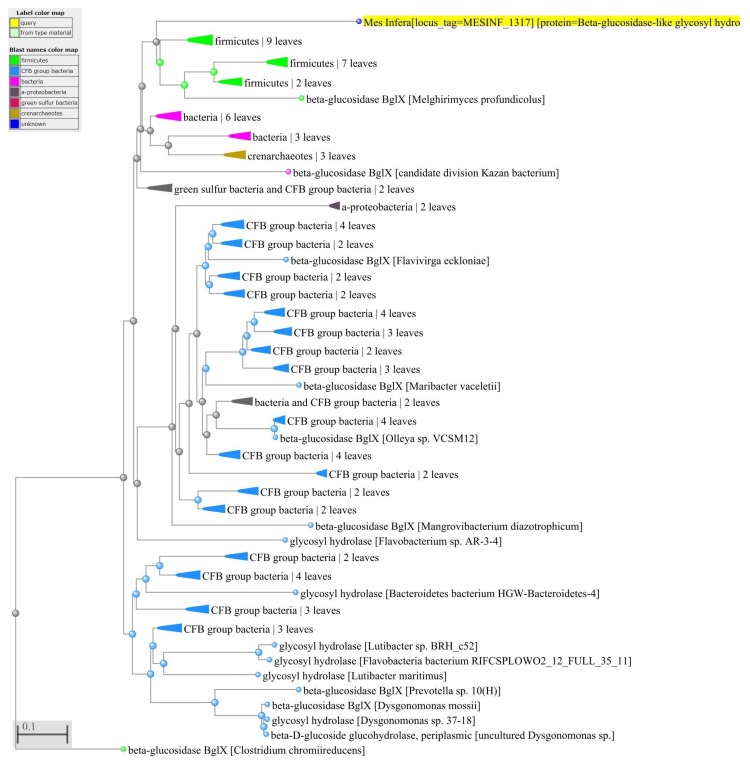
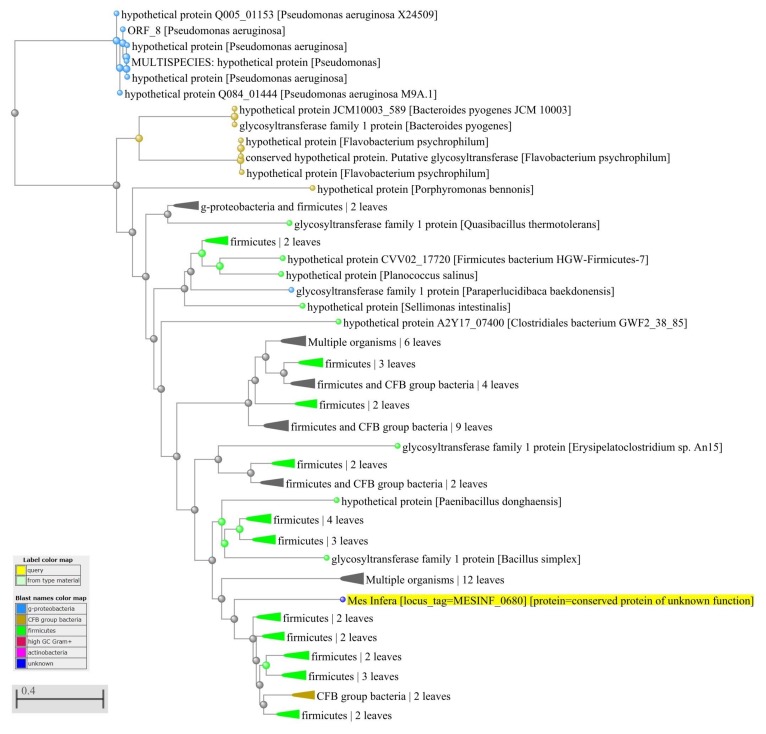
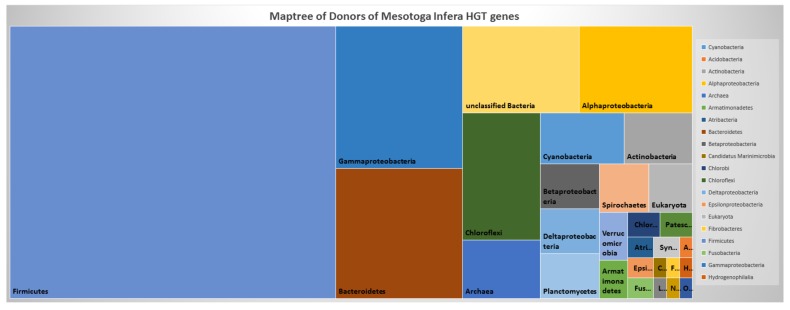
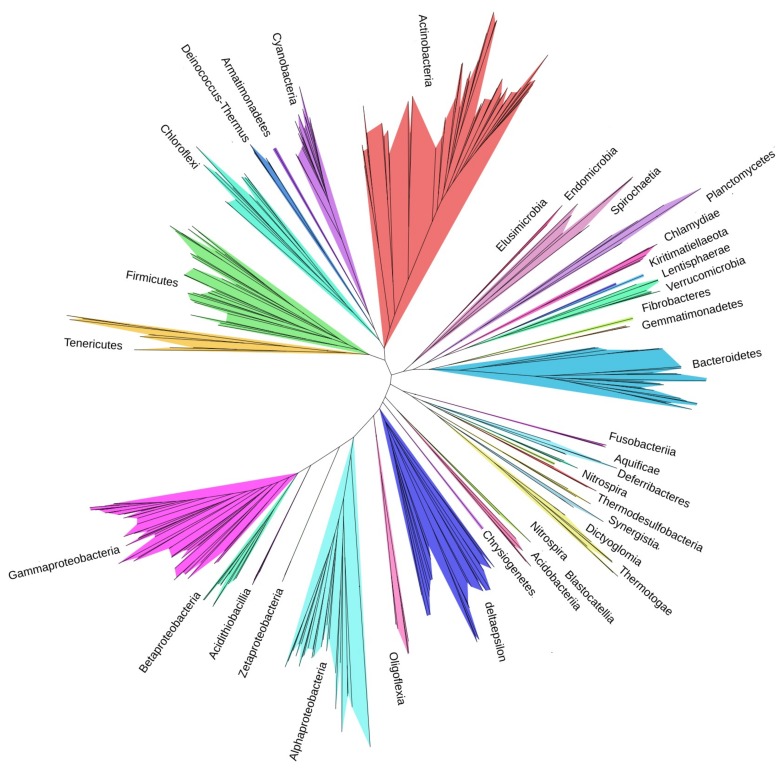
Reconstructing and visualizing phylogenetic relationships among living organisms is a fundamental challenge because not all organisms share the same genes. As a result, the first phylogenetic visualizations employed a single gene, e.g., rRNA genes, sufficiently conserved to be present in all organisms but divergent enough to provide discrimination between groups. As more genome data became available, researchers began concatenating different combinations of genes or proteins to construct phylogenetic trees believed to be more robust because they incorporated more information. However, the genes or proteins chosen were based on ad hoc approaches. The large number of complete genome sequences available today allows the use of whole genomes to analyze relationships among organisms rather than using an ad hoc set of genes. We present a systematic approach for constructing a phylogenetic tree based on simultaneously clustering the complete proteomes of 360 bacterial species. From the homologous clusters, we identify 49 protein sequences shared by 99% of the organisms to build a tree. Of the 49 sequences, 47 have homologous sequences in both archaea and eukarya. The clusters are also used to create a network from which bacterial species with horizontally-transferred genes from other phyla are identified.
重建并可视化生物之间的系统发育关系是一项根本性挑战,因为并非所有生物都拥有相同的基因。因此,最初的系统发育可视化采用单个基因,例如rRNA基因,其保守性足以存在于所有生物中,但又具有足够的差异性以区分不同的类群。随着越来越多的基因组数据可用,研究人员开始串联不同的基因或蛋白质组合来构建系统发育树,他们认为这样的树更可靠,因为纳入了更多信息。然而,所选择的基因或蛋白质是基于临时方法。如今大量完整的基因组序列使得可以使用全基因组来分析生物之间的关系,而不是使用一组临时选定的基因。我们提出了一种系统方法,基于对360种细菌物种的完整蛋白质组进行同时聚类来构建系统发育树。从同源簇中,我们鉴定出99%的生物共有的49个蛋白质序列来构建一棵树。在这49个序列中,47个在古细菌和真核生物中都有同源序列。这些簇还用于创建一个网络,从中识别出具有从其他门类水平转移基因的细菌物种。