Kamencek Tomas, Wieser Sandro, Kojima Hirotaka, Bedoya-Martínez Natalia, Dürholt Johannes P, Schmid Rochus, Zojer Egbert
Institute of Solid State Physics, Graz University of Technology, NAWI Graz, Petersgasse 16, 8010 Graz, Austria.
Institute of Physical and Theoretical Chemistry, Graz University of Technology, NAWI Graz, Stremayrgasse 9, 8010 Graz, Austria.
J Chem Theory Comput. 2020 Apr 14;16(4):2716-2735. doi: 10.1021/acs.jctc.0c00119. Epub 2020 Mar 24.
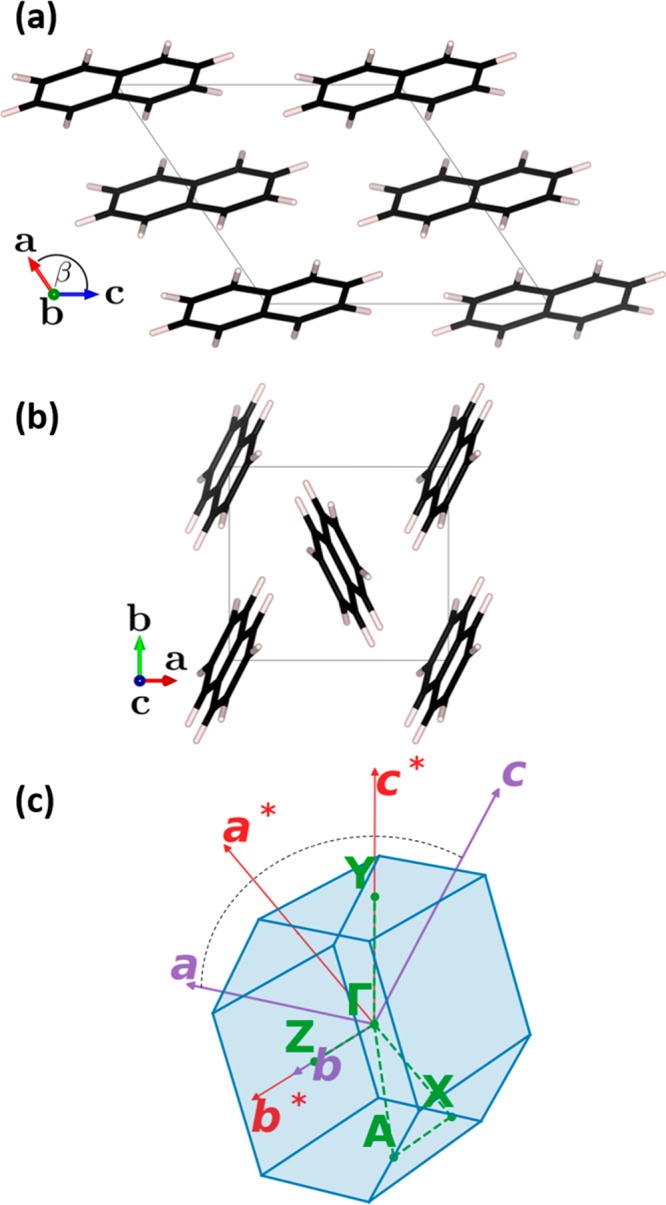
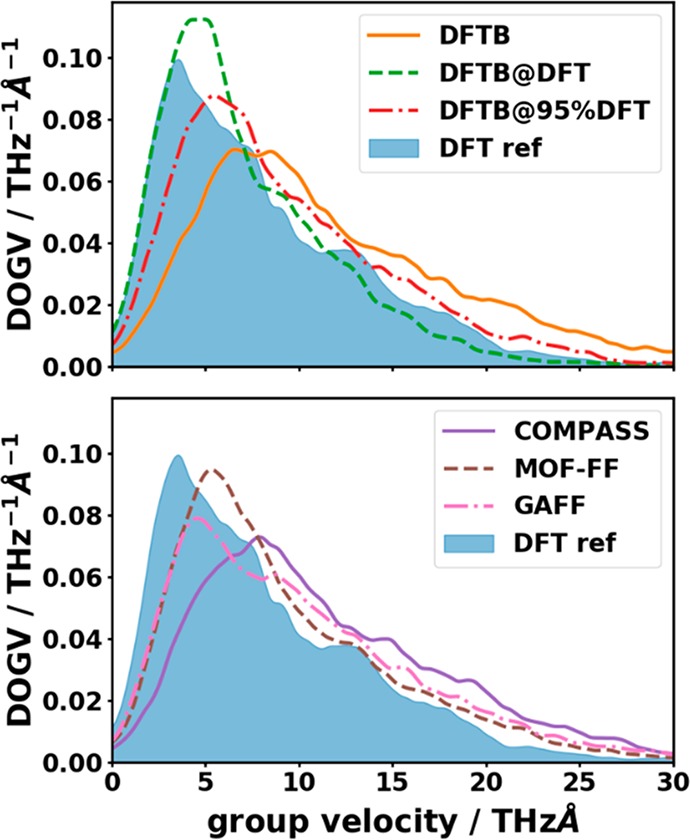
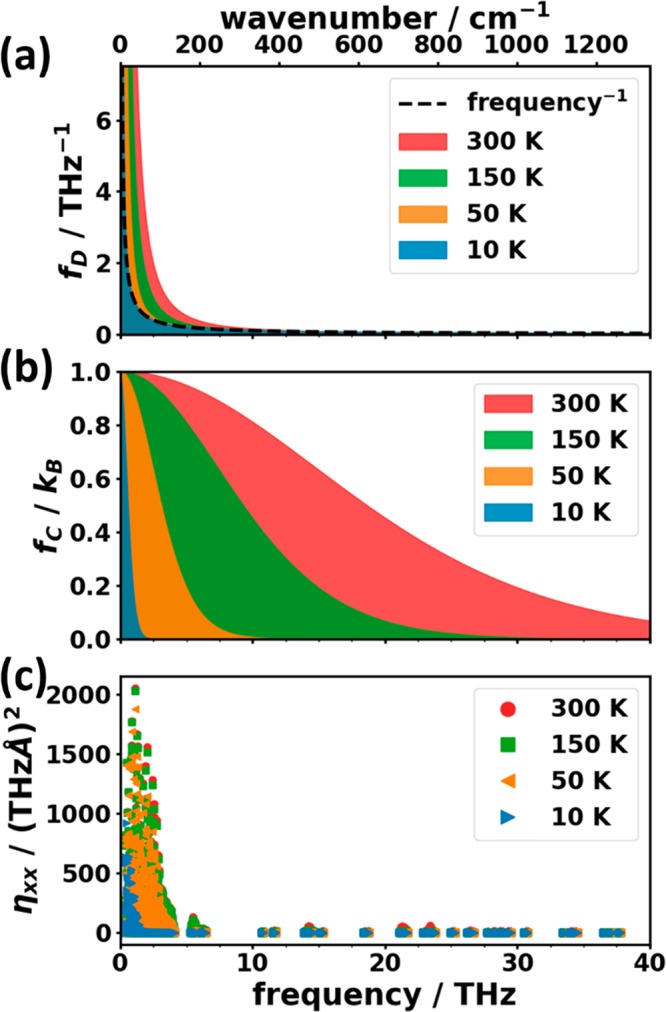
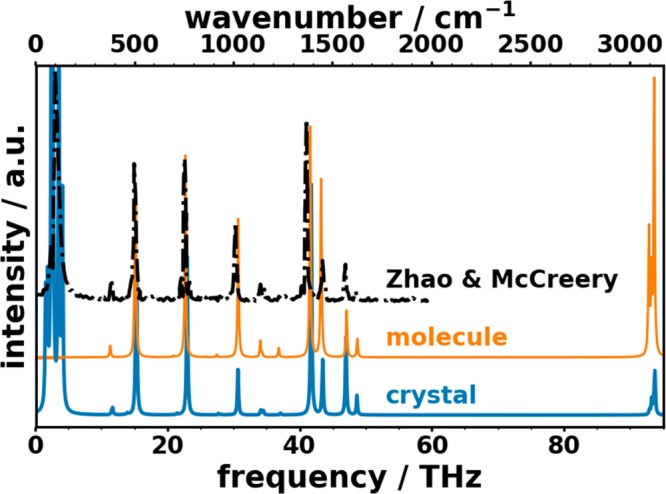
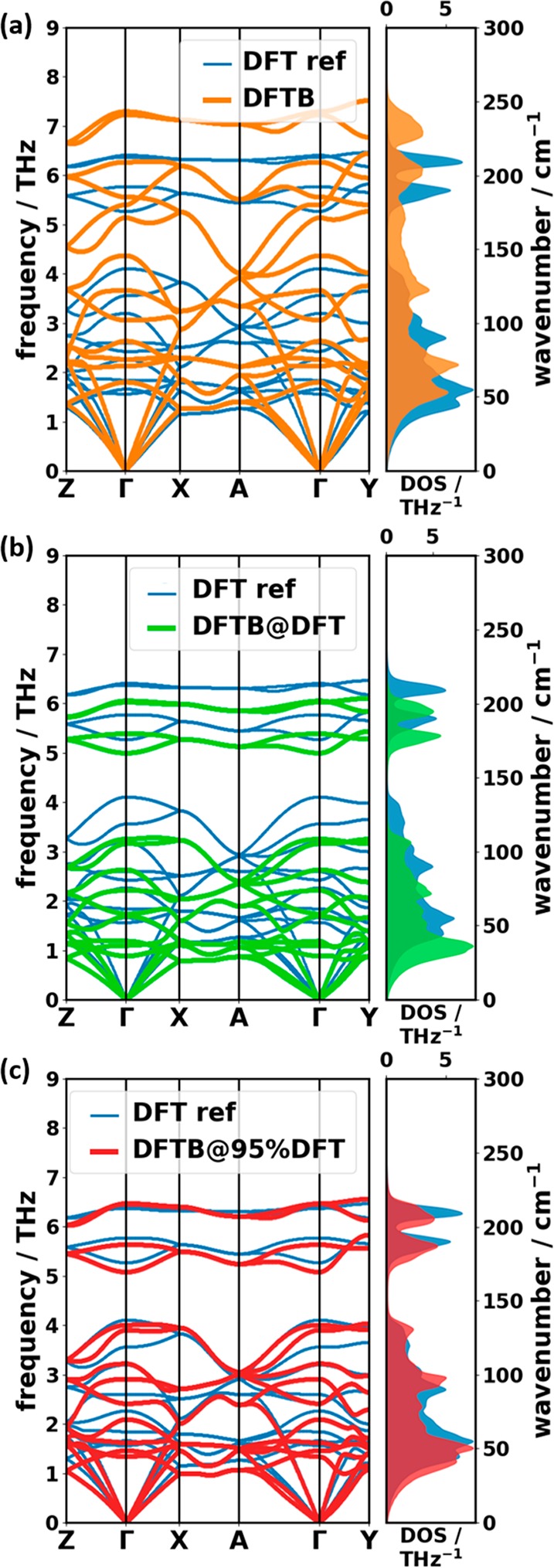
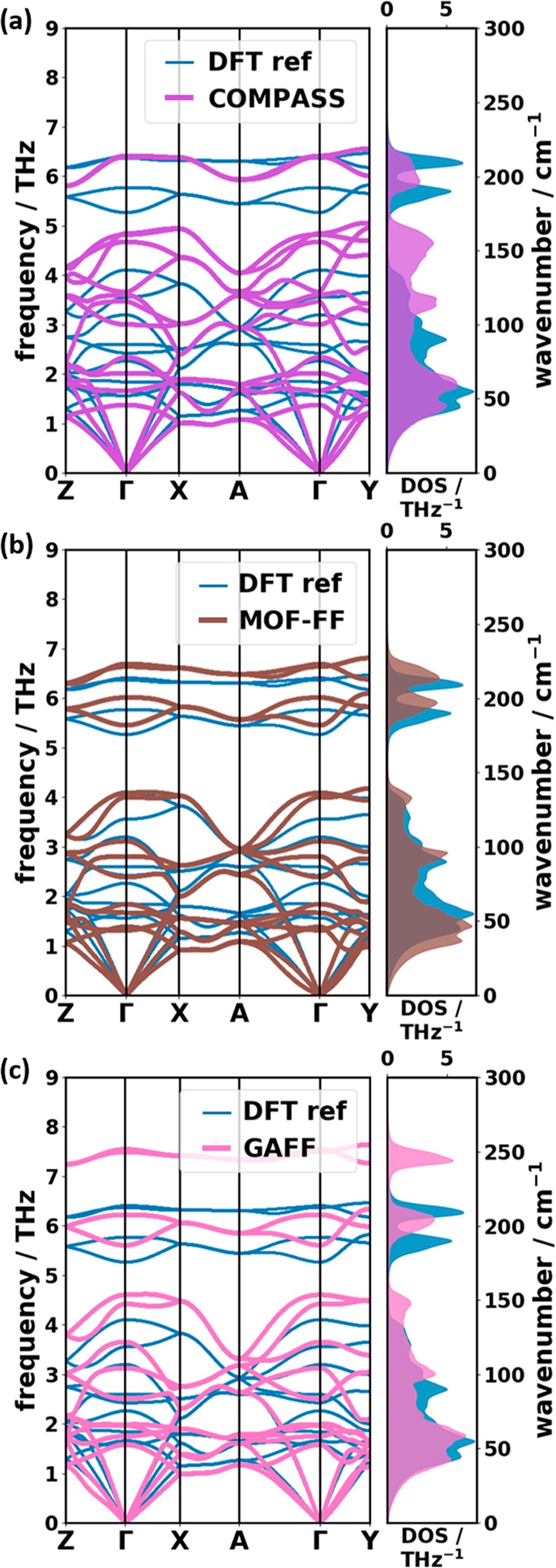
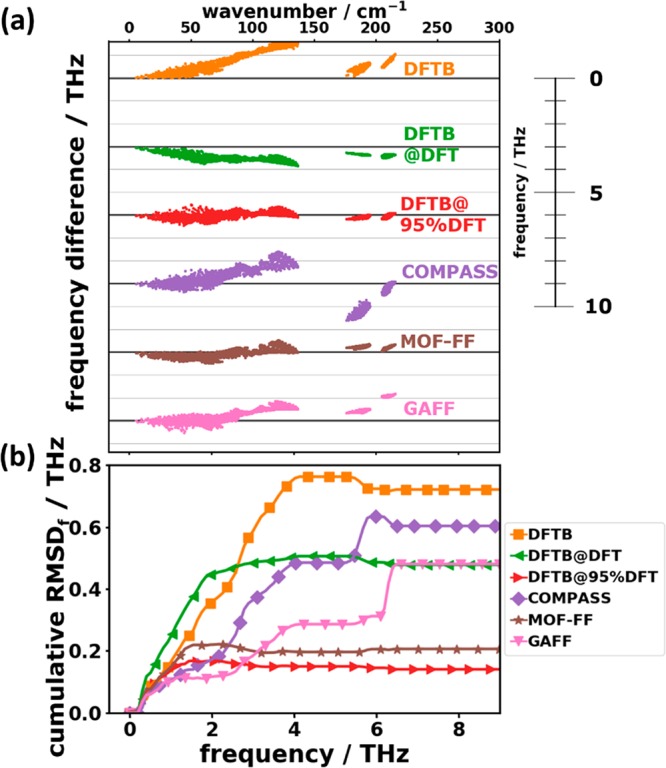
Phonons crucially impact a variety of properties of organic semiconductor materials. For instance, charge- and heat transport depend on low-frequency phonons, while for other properties, such as the free energy, especially high-frequency phonons count. For all these quantities one needs to know the entire phonon band structure, whose simulation becomes exceedingly expensive for more complex systems when using methods like dispersion-corrected density functional theory (DFT). Therefore, in the present contribution we evaluate the performance of more approximate methodologies, including density functional tight binding (DFTB) and a pool of force fields (FF) of varying complexity and sophistication. Beyond merely comparing phonon band structures, we also critically evaluate to what extent derived quantities, like temperature-dependent heat capacities, mean squared thermal displacements, and temperature-dependent free energies are impacted by shortcomings in the description of the phonon bands. As a benchmark system, we choose (deuterated) naphthalene, as the only organic semiconductor material for which to date experimental phonon band structures are available in the literature. Overall, the best performance among the approximate methodologies is observed for a system-specifically parametrized second-generation force field. Interestingly, in the low-frequency regime also force fields with a rather simplistic model for the bonding interactions (like the General Amber Force Field) perform rather well. As far as the tested DFTB parametrization is concerned, we obtain a significant underestimation of the unit-cell volume resulting in a pronounced overestimation of the phonon energies in the low-frequency region. This cannot be mended by relying on the DFT-calculated unit cell, since with this unit cell the DFTB phonon frequencies significantly underestimate the experiments.
声子对有机半导体材料的多种性质有着至关重要的影响。例如,电荷传输和热传输取决于低频声子,而对于其他性质,如自由能,则尤其依赖高频声子。对于所有这些量,人们需要了解整个声子能带结构,而当使用诸如色散校正密度泛函理论(DFT)等方法时,对于更复杂的系统,其模拟成本会变得极高。因此,在本论文中,我们评估了更近似方法的性能,包括密度泛函紧束缚(DFTB)以及一系列复杂度和精细程度各异的力场(FF)。除了仅仅比较声子能带结构外,我们还严格评估了诸如与温度相关的热容、均方热位移以及与温度相关的自由能等导出量在多大程度上受到声子能带描述缺陷的影响。作为一个基准系统,我们选择(氘代)萘,因为它是文献中目前唯一有实验声子能带结构的有机半导体材料。总体而言,在近似方法中,针对特定系统参数化的第二代力场表现最佳。有趣的是,在低频区域,对于键合相互作用采用相当简单模型的力场(如通用琥珀力场)也表现良好。就所测试的DFTB参数化而言,我们发现其显著低估了晶胞体积,导致在低频区域声子能量被明显高估。依靠DFT计算的晶胞并不能解决这个问题,因为使用这个晶胞时,DFTB声子频率会显著低估实验值。