Institute of Molecular Cell Biology, Center for Molecular Biomedicine, Jena University Hospital, 07743 Jena, Germany.
Institute of Biochemistry II and Center for Sepsis Control and Care, Jena University Hospital, 07743 Jena, Germany.
Cells. 2020 Mar 11;9(3):687. doi: 10.3390/cells9030687.
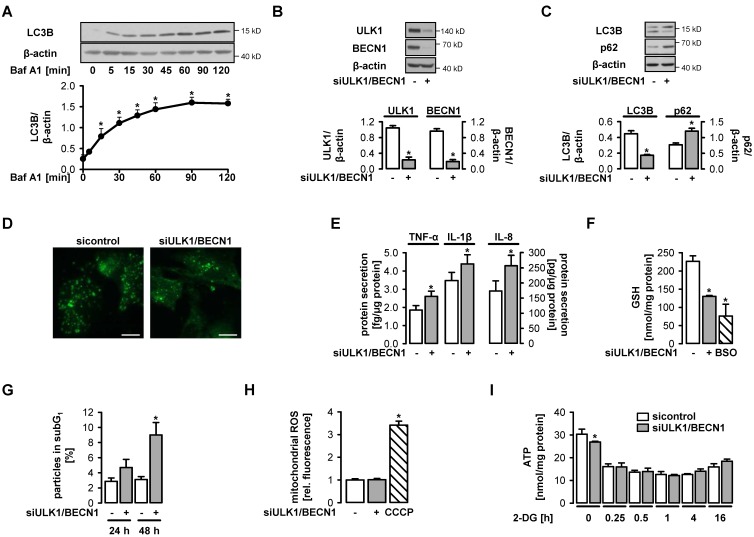
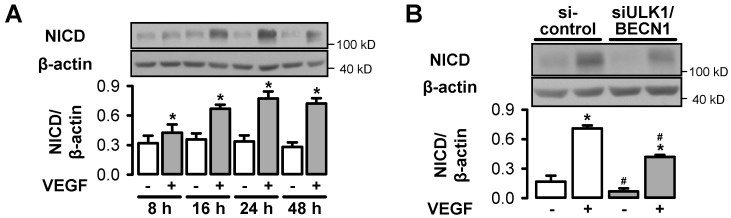
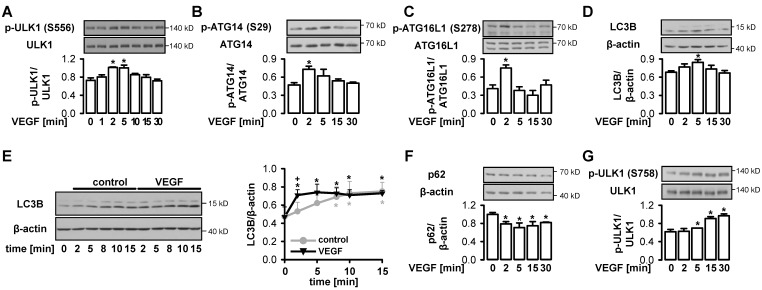
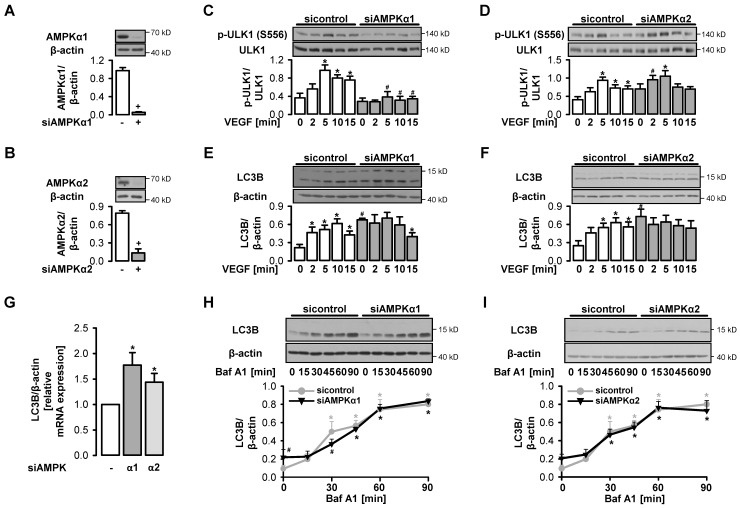
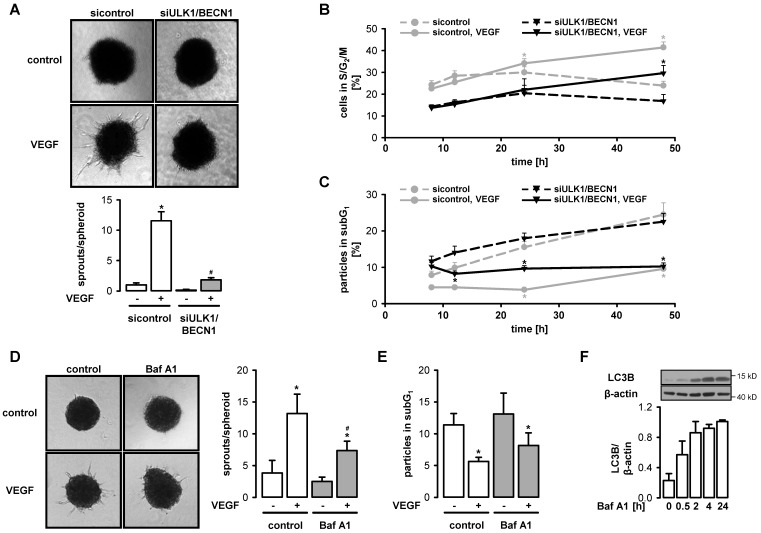
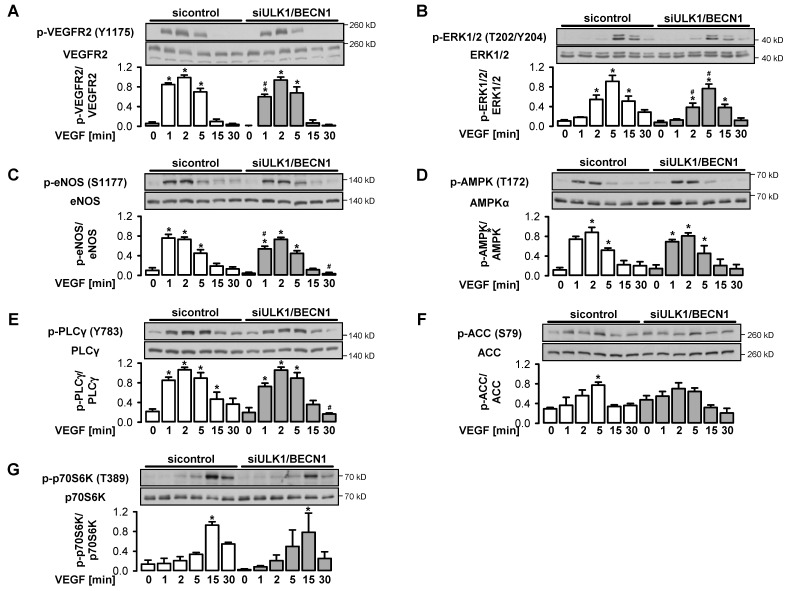
AMP-activated protein kinase (AMPK) is activated by vascular endothelial growth factor (VEGF) in endothelial cells and it is significantly involved in VEGF-induced angiogenesis. This study investigates whether the VEGF/AMPK pathway regulates autophagy in endothelial cells and whether this is linked to its pro-angiogenic role. We show that VEGF leads to AMPKα1-dependent phosphorylation of Unc-51-like kinase 1 (ULK1) at its serine residue 556 and to the subsequent phosphorylation of the ULK1 substrate ATG14. This triggers initiation of autophagy as shown by phosphorylation of ATG16L1 and conjugation of the microtubule-associated protein light chain 3B, which indicates autophagosome formation; this is followed by increased autophagic flux measured in the presence of bafilomycin A1 and by reduced expression of the autophagy substrate p62. VEGF-induced autophagy is transient and probably terminated by mechanistic target of rapamycin (mTOR), which is activated by VEGF in a delayed manner. We show that functional autophagy is required for VEGF-induced angiogenesis and may have specific functions in addition to maintaining homeostasis. In line with this, inhibition of autophagy impaired VEGF-mediated formation of the Notch intracellular domain, a critical regulator of angiogenesis. Our study characterizes autophagy induction as a pro-angiogenic function of the VEGF/AMPK pathway and suggests that timely activation of autophagy-initiating pathways may help to initiate angiogenesis.
腺苷酸活化蛋白激酶 (AMPK) 在血管内皮细胞中被血管内皮生长因子 (VEGF) 激活,它在 VEGF 诱导的血管生成中起着重要作用。本研究探讨了 VEGF/AMPK 通路是否调节内皮细胞中的自噬,以及这是否与其促血管生成作用有关。我们发现,VEGF 导致 AMPKα1 依赖性地将 Unc-51 样激酶 1 (ULK1) 的丝氨酸残基 556 磷酸化,并随后磷酸化 ULK1 底物 ATG14。这触发了自噬的起始,如 ATG16L1 的磷酸化和微管相关蛋白轻链 3B 的缀合所表明的,这表明自噬体的形成;随后在存在巴弗洛霉素 A1 时测量到自噬通量增加,并通过自噬底物 p62 的表达减少来测量。VEGF 诱导的自噬是短暂的,可能被机械性靶标雷帕霉素 (mTOR) 终止,mTOR 被 VEGF 延迟激活。我们表明,功能性自噬是 VEGF 诱导的血管生成所必需的,并且除了维持内稳态之外,可能具有特定的功能。与此一致,自噬的抑制损害了 VEGF 介导的 Notch 细胞内结构域的形成,Notch 细胞内结构域是血管生成的关键调节剂。我们的研究将自噬诱导特征化为 VEGF/AMPK 通路的促血管生成功能,并表明及时激活自噬起始途径可能有助于启动血管生成。