Plant Ewan P, Manukyan Hasmik, Sanchez Jose L, Laassri Majid, Ye Zhiping
Division of Viral Products, Center for Biologics Evaluation and Research, US Food and Drug Administration, Silver Spring, MD 20993, USA.
Armed Forces Health Surveillance Branch, Public Health Division, Assistant Director for Combat Support (AD-CS), Defense Health Agency, Silver Spring, MD 20904, USA.
Vaccines (Basel). 2020 Mar 11;8(1):125. doi: 10.3390/vaccines8010125.
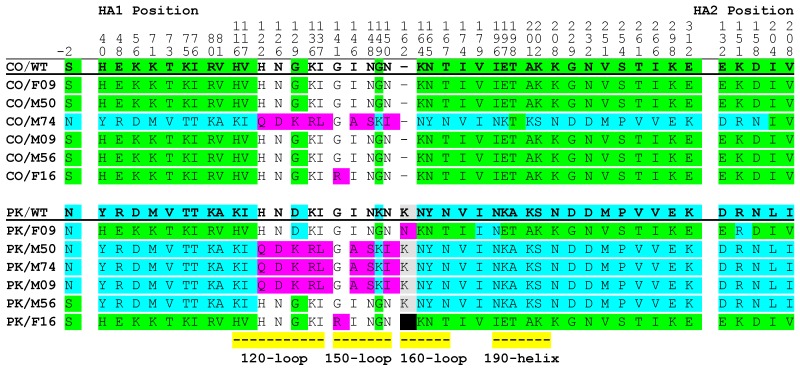
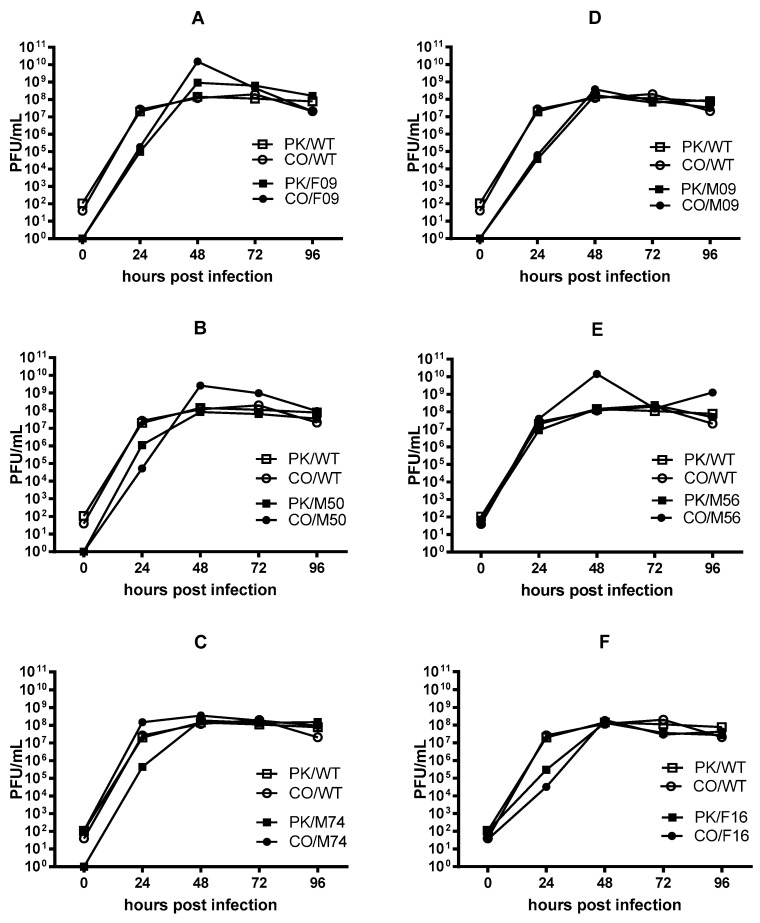
Mutations arise in the genomes of progeny viruses during infection. Mutations that occur in epitopes targeted by host antibodies allow the progeny virus to escape the host adaptive, B-cell mediated antibody immune response. Major epitopes have been identified in influenza B virus (IBV) hemagglutinin (HA) protein. However, IBV strains maintain a seasonal presence in the human population and changes in IBV genomes in response to immune pressure are not well characterized. There are two lineages of IBV that have circulated in the human population since the 1980s, B-Victoria and B-Yamagata. It is hypothesized that early exposure to one influenza subtype leads to immunodominance. Subsequent seasonal vaccination or exposure to new subtypes may modify subsequent immune responses, which, in turn, results in selection of escape mutations in the viral genome. Here we show that while some mutations do occur in known epitopes suggesting antibody escape, many mutations occur in other parts of the HA protein. Analysis of mutations outside of the known epitopes revealed that these mutations occurred at the same amino acid position in viruses from each of the two IBV lineages. Interestingly, where the amino acid sequence differed between viruses from each lineage, reciprocal amino acid changes were observed. That is, the virus from the Yamagata lineage become more like the Victoria lineage virus and vice versa. Our results suggest that some IBV HA sequences are constrained to specific amino acid codons when viruses are cultured in the presence of antibodies. Some changes to the known antigenic regions may also be restricted in a lineage-dependent manner. Questions remain regarding the mechanisms underlying these results. The presence of amino acid residues that are constrained within the HA may provide a new target for universal vaccines for IBV.
在感染过程中,子代病毒的基因组会出现突变。发生在宿主抗体靶向表位的突变使子代病毒能够逃避宿主适应性的、B细胞介导的抗体免疫反应。在乙型流感病毒(IBV)血凝素(HA)蛋白中已鉴定出主要表位。然而,IBV毒株在人群中呈季节性存在,且针对免疫压力的IBV基因组变化尚未得到充分表征。自20世纪80年代以来,有两种IBV谱系在人群中传播,即B-维多利亚系和B-山形系。据推测,早期接触一种流感亚型会导致免疫优势。随后的季节性疫苗接种或接触新亚型可能会改变后续的免疫反应,进而导致病毒基因组中逃逸突变的选择。在这里,我们表明,虽然一些突变确实发生在已知表位中,提示抗体逃逸,但许多突变发生在HA蛋白的其他部位。对已知表位以外的突变分析表明,这些突变发生在来自两个IBV谱系中每个谱系的病毒的相同氨基酸位置。有趣的是,当每个谱系的病毒氨基酸序列不同时,观察到了相互的氨基酸变化。也就是说,山形谱系的病毒变得更像维多利亚谱系的病毒,反之亦然。我们的结果表明,当病毒在抗体存在的情况下培养时,一些IBV HA序列被限制在特定的氨基酸密码子上。已知抗原区域的一些变化也可能以谱系依赖的方式受到限制。关于这些结果背后的机制仍存在疑问。HA中受限制的氨基酸残基的存在可能为IBV通用疫苗提供新的靶点。