Liu Yansheng, Gonzàlez-Porta Mar, Santos Sergio, Brazma Alvis, Marioni John C, Aebersold Ruedi, Venkitaraman Ashok R, Wickramasinghe Vihandha O
Department of Biology, Institute of Molecular Systems Biology, ETH Zurich, Zurich, Switzerland.
European Molecular Biology Laboratory-European Bioinformatics Institute (EMBL-EBI), Hinxton, UK.
Cell Rep. 2017 Aug 1;20(5):1229-1241. doi: 10.1016/j.celrep.2017.07.025.
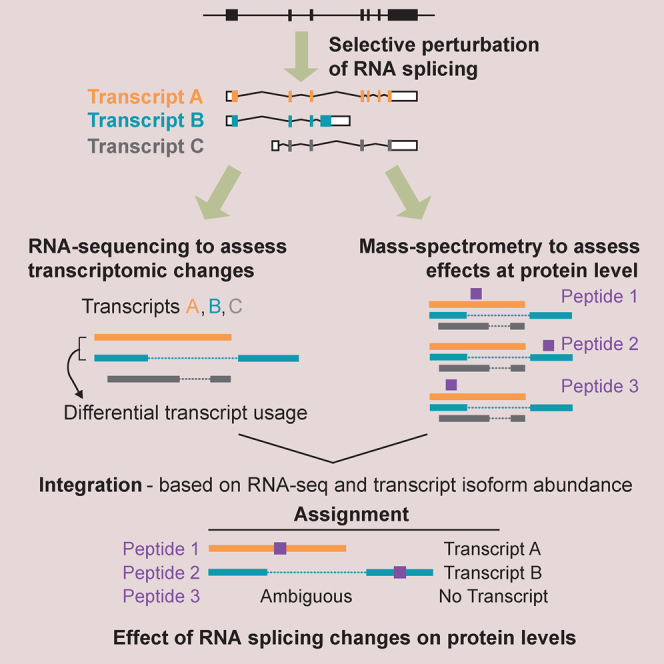
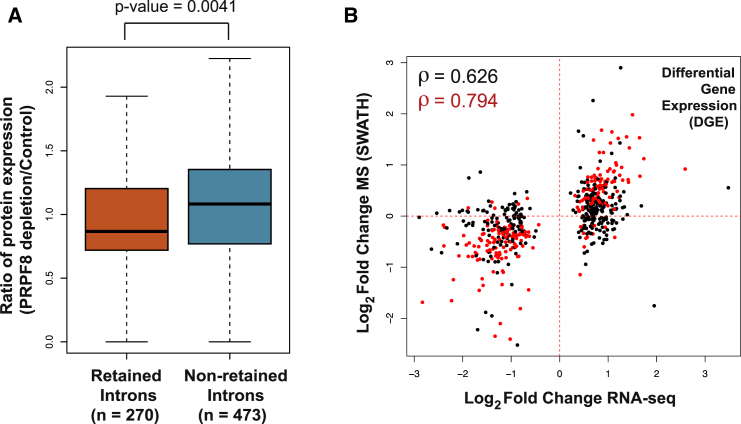
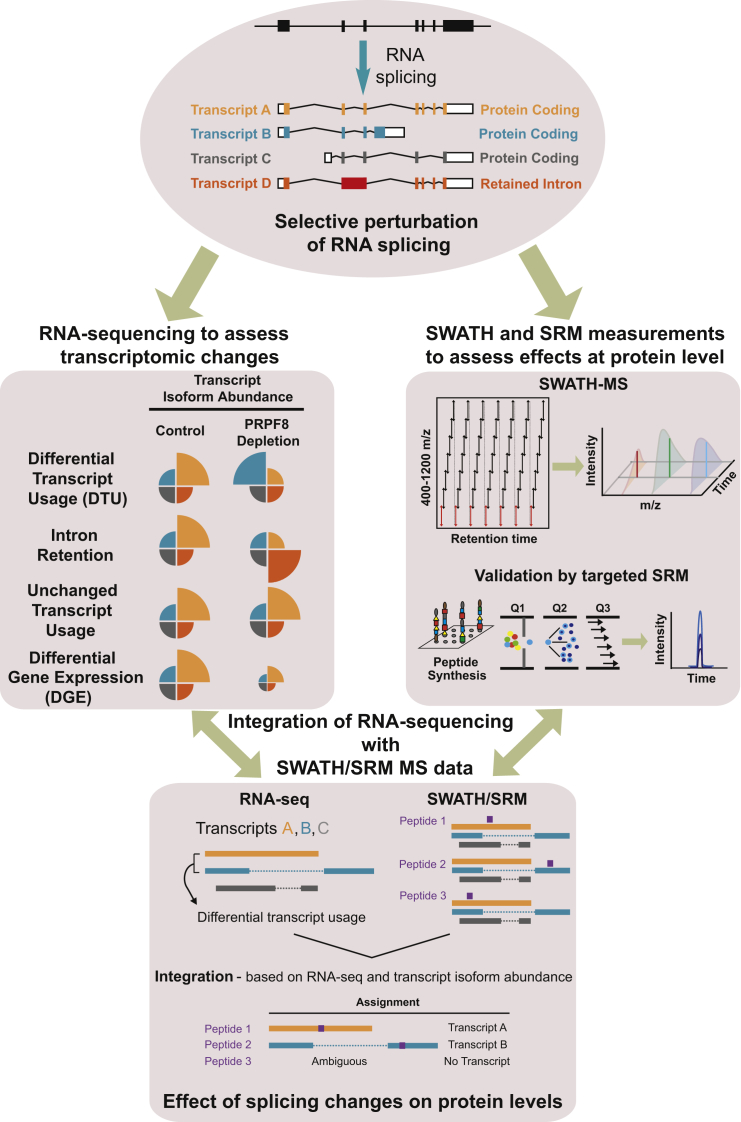
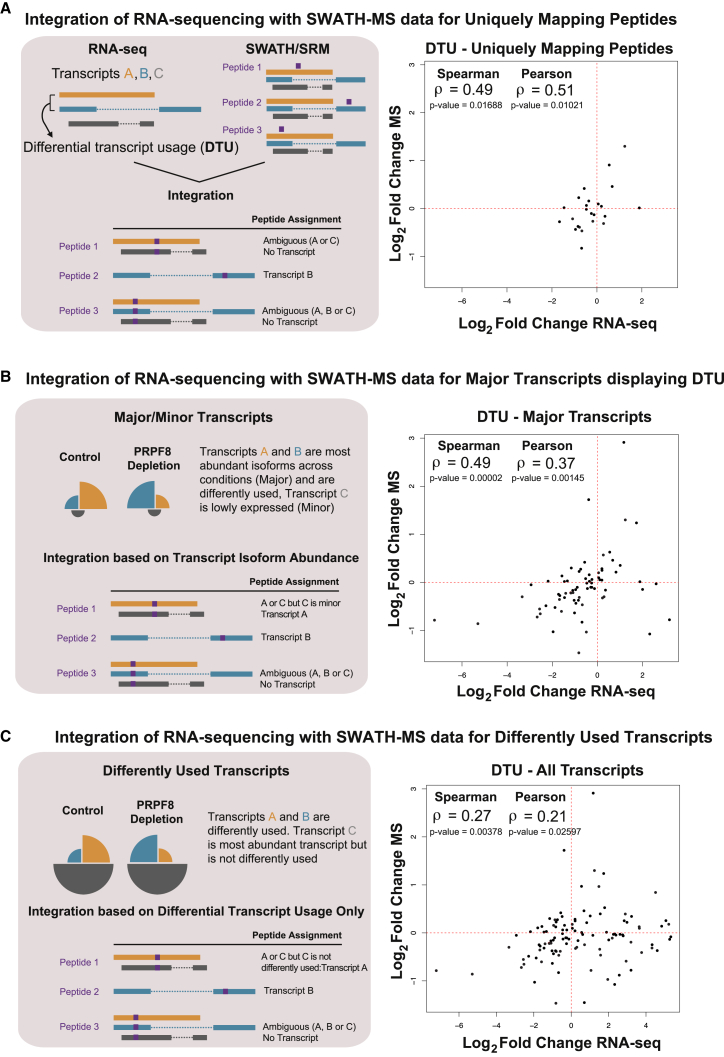
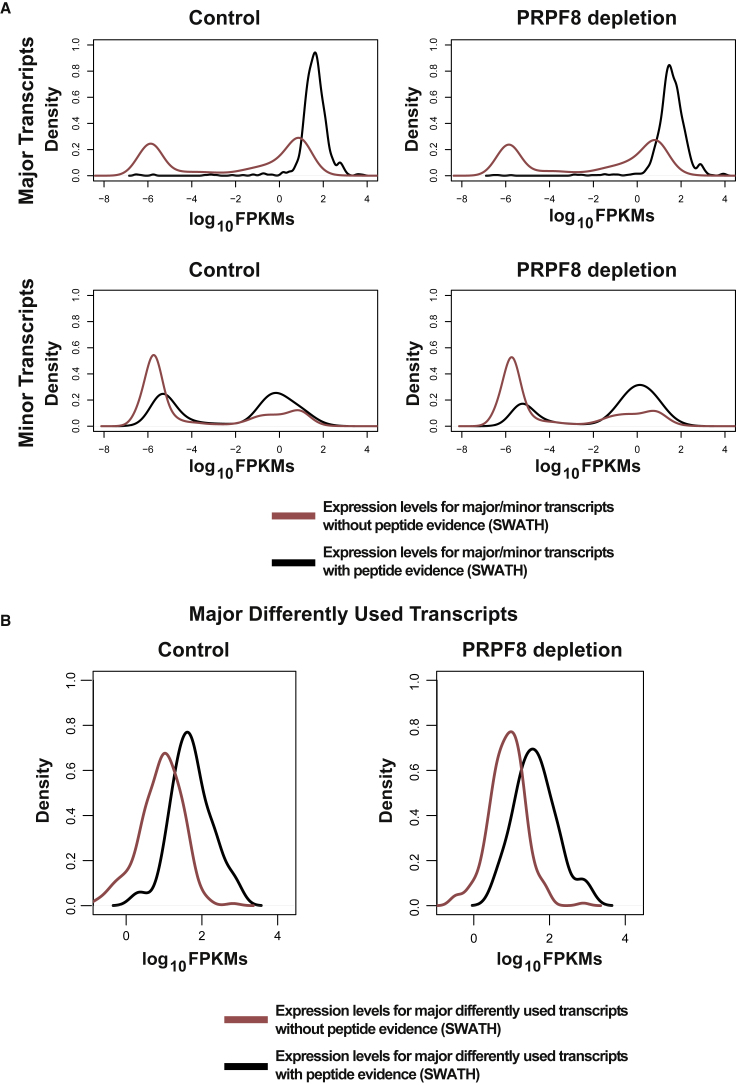
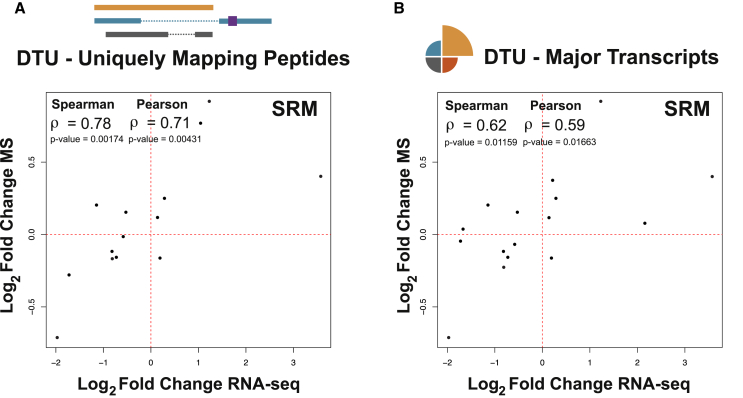
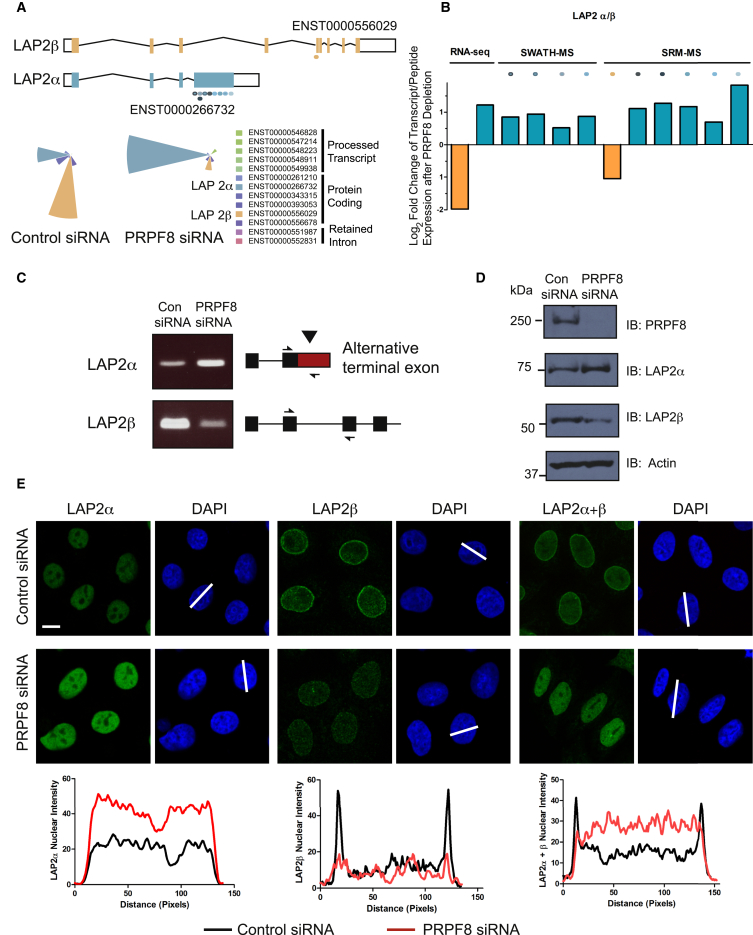
Alternative splicing is a critical determinant of genome complexity and, by implication, is assumed to engender proteomic diversity. This notion has not been experimentally tested in a targeted, quantitative manner. Here, we have developed an integrative approach to ask whether perturbations in mRNA splicing patterns alter the composition of the proteome. We integrate RNA sequencing (RNA-seq) (to comprehensively report intron retention, differential transcript usage, and gene expression) with a data-independent acquisition (DIA) method, SWATH-MS (sequential window acquisition of all theoretical spectra-mass spectrometry), to capture an unbiased, quantitative snapshot of the impact of constitutive and alternative splicing events on the proteome. Whereas intron retention is accompanied by decreased protein abundance, alterations in differential transcript usage and gene expression alter protein abundance proportionate to transcript levels. Our findings illustrate how RNA splicing links isoform expression in the human transcriptome with proteomic diversity and provides a foundation for studying perturbations associated with human diseases.
可变剪接是基因组复杂性的关键决定因素,因此被认为会产生蛋白质组多样性。这一概念尚未以有针对性的定量方式进行实验验证。在这里,我们开发了一种综合方法,以探究mRNA剪接模式的扰动是否会改变蛋白质组的组成。我们将RNA测序(RNA-seq)(用于全面报告内含子保留、差异转录本使用情况和基因表达)与一种数据非依赖采集(DIA)方法——SWATH-MS(所有理论谱图的顺序窗口采集-质谱分析法)相结合,以获取组成型和可变剪接事件对蛋白质组影响的无偏定量快照。内含子保留伴随着蛋白质丰度的降低,而差异转录本使用情况和基因表达的改变则会使蛋白质丰度与转录本水平成比例地变化。我们的研究结果阐明了RNA剪接如何将人类转录组中的异构体表达与蛋白质组多样性联系起来,并为研究与人类疾病相关的扰动提供了基础。