Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawinskiego 5a, 02-106, Warsaw, Poland.
Department of Systems Biology, Institute of Experimental Plant Biology and Biotechnology, University of Warsaw, Miecznikowa 1, 02-096, Warsaw, Poland.
Sci Rep. 2020 Mar 19;10(1):5035. doi: 10.1038/s41598-020-61860-z.
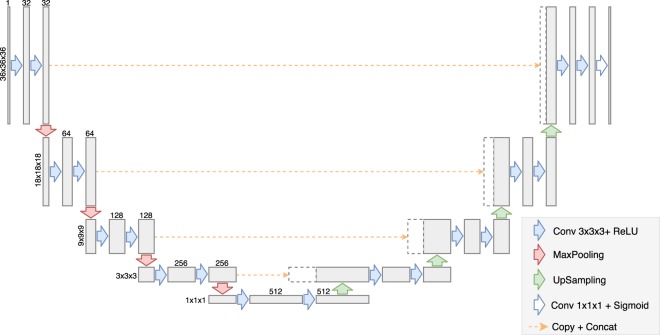
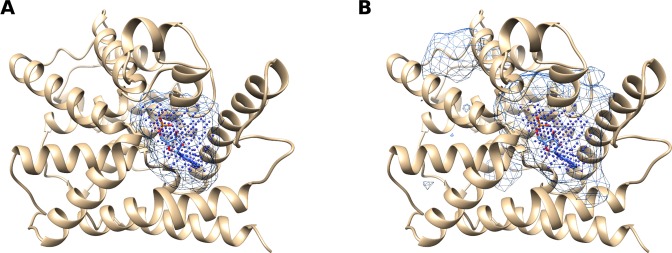
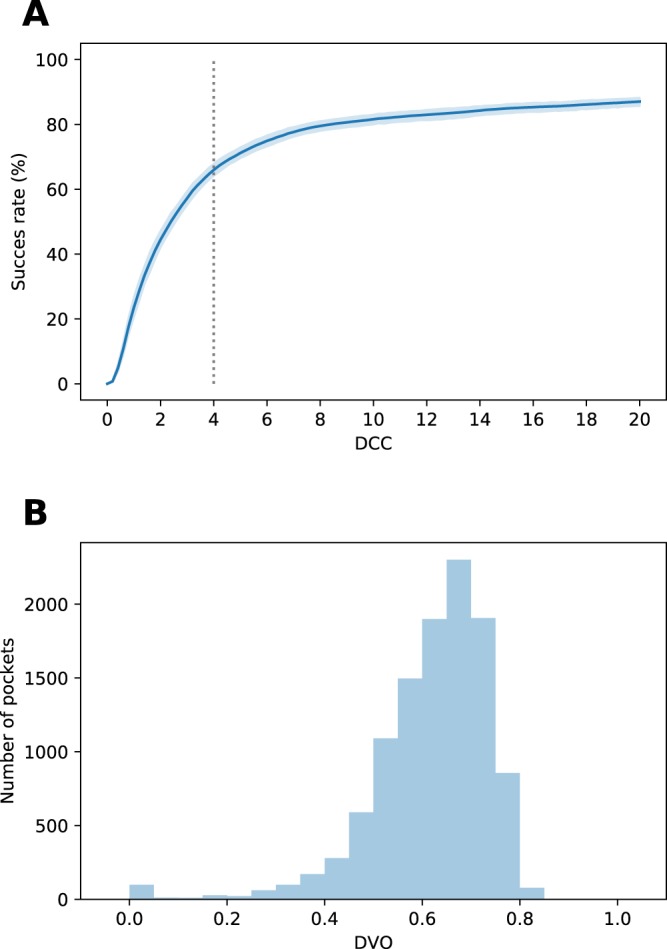
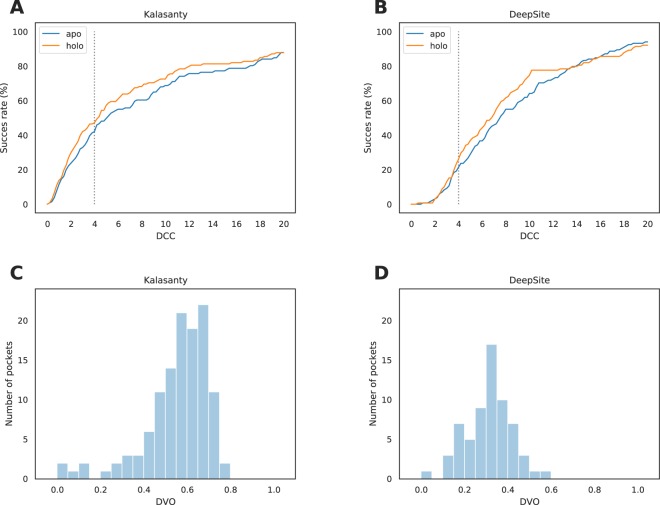
In recent years machine learning (ML) took bio- and cheminformatics fields by storm, providing new solutions for a vast repertoire of problems related to protein sequence, structure, and interactions analysis. ML techniques, deep neural networks especially, were proven more effective than classical models for tasks like predicting binding affinity for molecular complex. In this work we investigated the earlier stage of drug discovery process - finding druggable pockets on protein surface, that can be later used to design active molecules. For this purpose we developed a 3D fully convolutional neural network capable of binding site segmentation. Our solution has high prediction accuracy and provides intuitive representations of the results, which makes it easy to incorporate into drug discovery projects. The model's source code, together with scripts for most common use-cases is freely available at http://gitlab.com/cheminfIBB/kalasanty.
近年来,机器学习(ML)在生物信息学和化学信息学领域掀起了一场革命,为与蛋白质序列、结构和相互作用分析相关的大量问题提供了新的解决方案。ML 技术,特别是深度神经网络,在预测分子复合物的结合亲和力等任务上被证明比传统模型更有效。在这项工作中,我们研究了药物发现过程的早期阶段——在蛋白质表面找到可成药的口袋,这些口袋可用于设计活性分子。为此,我们开发了一个能够进行结合位点分割的 3D 全卷积神经网络。我们的解决方案具有很高的预测准确性,并提供了直观的结果表示,这使得它易于纳入药物发现项目。该模型的源代码以及最常见用例的脚本可在 http://gitlab.com/cheminfIBB/kalasanty 上免费获得。