Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw, Poland.
Department of Systems Biology, Institute of Experimental Plant Biology and Biotechnology, University of Warsaw, Warsaw, Poland.
Bioinformatics. 2018 Nov 1;34(21):3666-3674. doi: 10.1093/bioinformatics/bty374.
Structure based ligand discovery is one of the most successful approaches for augmenting the drug discovery process. Currently, there is a notable shift towards machine learning (ML) methodologies to aid such procedures. Deep learning has recently gained considerable attention as it allows the model to 'learn' to extract features that are relevant for the task at hand.
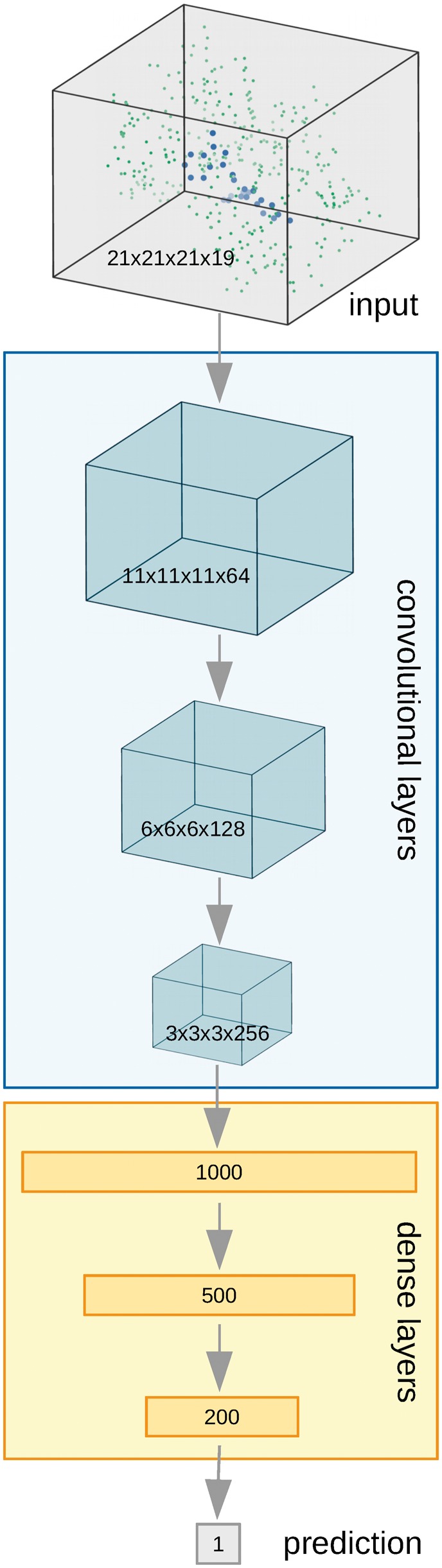
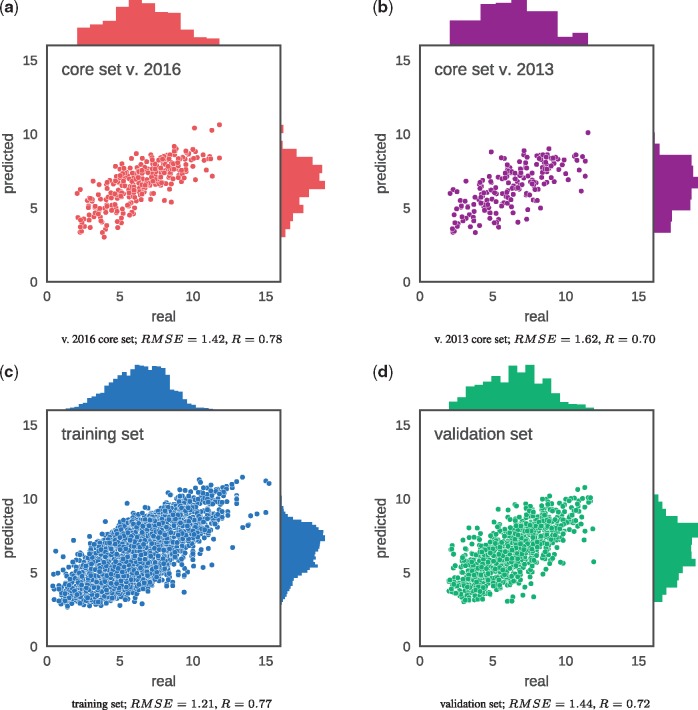
We have developed a novel deep neural network estimating the binding affinity of ligand-receptor complexes. The complex is represented with a 3D grid, and the model utilizes a 3D convolution to produce a feature map of this representation, treating the atoms of both proteins and ligands in the same manner. Our network was tested on the CASF-2013 'scoring power' benchmark and Astex Diverse Set and outperformed classical scoring functions.
The model, together with usage instructions and examples, is available as a git repository at http://gitlab.com/cheminfIBB/pafnucy.
Supplementary data are available at Bioinformatics online.
基于结构的配体发现是增强药物发现过程最成功的方法之一。目前,机器学习 (ML) 方法在辅助这些过程方面有明显的转变。深度学习最近引起了相当大的关注,因为它允许模型“学习”提取与手头任务相关的特征。
我们开发了一种新的深度神经网络,用于估计配体-受体复合物的结合亲和力。复合物用 3D 网格表示,模型利用 3D 卷积来生成此表示的特征图,以相同的方式对待蛋白质和配体的原子。我们的网络在 CASF-2013“评分能力”基准和 Astex 多样集上进行了测试,性能优于经典评分函数。
该模型以及使用说明和示例可作为 git 存储库在 http://gitlab.com/cheminfIBB/pafnucy 上获得。
补充数据可在 Bioinformatics 在线获得。