Beaudin Marie, Sellami Leila, Martel Christian, Touzel-Deschênes Lydia, Houle Gabrielle, Martineau Laurence, Lacroix Kevin, Lavallée Andréane, Chrestian Nicolas, Rouleau Guy A, Gros-Louis François, Laforce Robert, Dupré Nicolas
Department of Medicine (M.B., L.S., N.D.), Faculty of Medicine, Université Laval; Division of Neurosciences (M.B., L.M., N.D.), CHU de Québec - Université Laval; Clinique Interdisciplinaire de Mémoire (L.S., R.L.), CHU de Québec; Laval University Experimental Organogenesis Research Center/LOEX (C.M., L.T.-D., F.G.-L.), Division of Regenerative Medicine, CHU de Québec Research Center - Enfant-Jésus Hospital; Montreal Neurological Institute (G.H., G.A.R.), McGill University, Québec, Canada; CHU Grenoble-Alpes (L.M.), Grenoble, France; CIUSSS de la Mauricie-et-du-Centre-du-Québec (K.L.), Trois-Rivières; Centre universitaire d'ophtalmologie (A.L.), Department of Surgery, Faculty of Medicine, CHU de Québec - Université Laval; and Centre Mère-Enfant-Soleil (N.C.), Université Laval, Québec, Canada.
Neurol Genet. 2020 Feb 20;6(2):e403. doi: 10.1212/NXG.0000000000000403. eCollection 2020 Apr.
To better characterize the neurologic and cognitive profile of patients with spinocerebellar ataxia 34 (SCA34) caused by mutations and to demonstrate the presence of ELOVL4 cellular localization and distribution abnormalities in skin-derived fibroblasts.
We investigated a 5-generation French-Canadian kindred presenting with a late-onset cerebellar ataxia and recruited age- and education-matched controls to evaluate the presence of neurocognitive impairment. Immunohistochemistry of dermal fibroblasts derived from a patient's skin biopsy was performed.
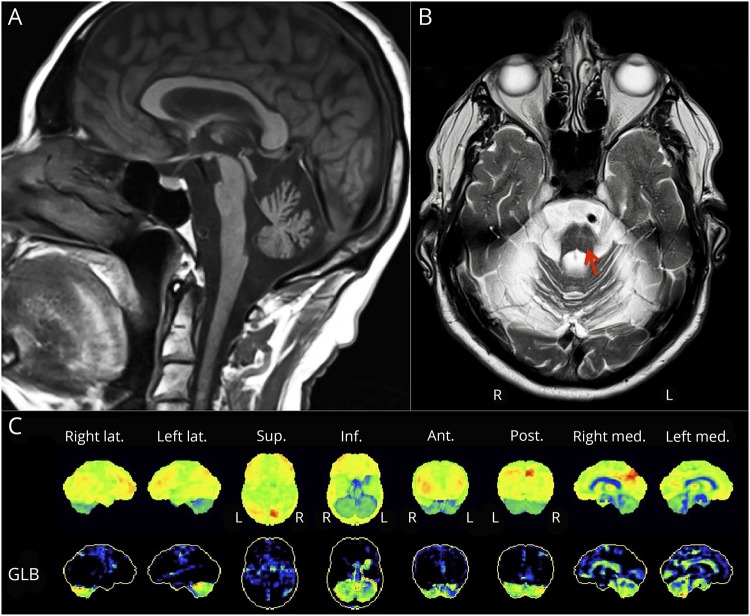
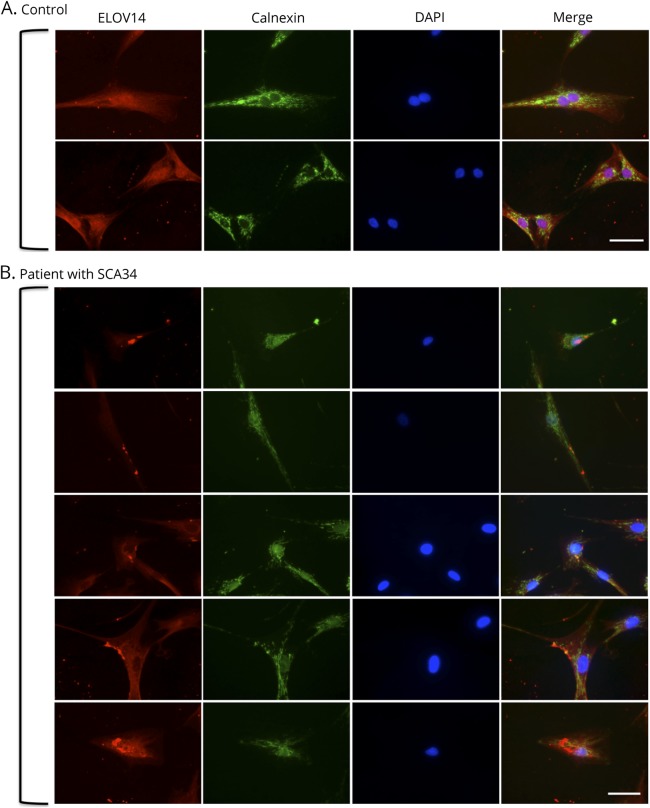
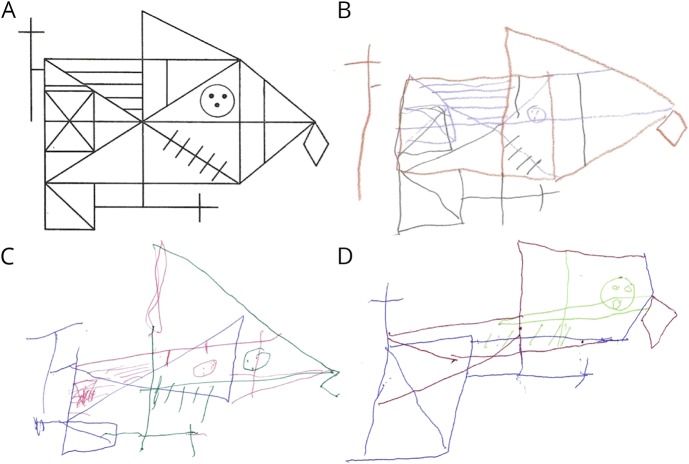
Patients had a late-onset slowly progressive cerebellar syndrome (mean age at onset 47 years; range 32-60 years) characterized by truncal and limb ataxia, dysarthria, hypometric saccades, and saccadic pursuits. No patient had past or current signs of erythrokeratodermia variabilis, which had previously been reported. MRI revealed cerebellar atrophy, with pontine atrophy (4 of 6 patients), and cruciform hypersignal in the pons (2 of 6 patients). Fluorodeoxyglucose-PET showed diffuse cerebellar hypometabolism in all 5 tested patients with subtle parietal hypometabolism in 3. Significant cognitive deficits were found in executive functioning, along with apparent visuospatial, attention, and psychiatric involvement. Immunohistochemistry of dermal fibroblasts showed mislocalization of the ELOVL4 protein, which appeared punctate and aggregated, supporting a dominant negative effect of the mutation on protein localization.
Our findings support the pathogenicity of mutations in cerebellar dysfunction and provide a detailed characterization of the SCA34 phenotype, with neurocognitive changes typical of the cerebellar cognitive-affective syndrome.
更好地描述由突变引起的脊髓小脑共济失调34型(SCA34)患者的神经和认知特征,并证明皮肤来源的成纤维细胞中ELOVL4细胞定位和分布异常的存在。
我们调查了一个患有迟发性小脑共济失调的五代法裔加拿大家族,并招募了年龄和教育程度匹配的对照组来评估神经认知障碍的存在。对患者皮肤活检获得的真皮成纤维细胞进行免疫组织化学检测。
患者表现为迟发性缓慢进展的小脑综合征(平均发病年龄47岁;范围32 - 60岁),特征为躯干和肢体共济失调、构音障碍、眼扫视幅度减小和扫视跟踪。没有患者有既往或当前可变型红斑角化病的体征,该病此前已有报道。MRI显示小脑萎缩,伴有脑桥萎缩(6例患者中的4例),以及脑桥十字形高信号(6例患者中的2例)。氟脱氧葡萄糖PET显示所有5例受试患者均有弥漫性小脑代谢减低,3例有轻微顶叶代谢减低。在执行功能方面发现显著的认知缺陷,同时明显存在视觉空间、注意力和精神方面的问题。真皮成纤维细胞的免疫组织化学显示ELOVL4蛋白定位错误,呈点状和聚集状,支持该突变对蛋白定位的显性负效应。
我们的研究结果支持突变在小脑功能障碍中的致病性,并提供了SCA34表型的详细特征,具有小脑认知 - 情感综合征典型的神经认知变化。