Key Laboratory of Metallurgical Emission Reduction & Resources Recycling (Anhui University of Technology), Ministry of Education, Ma'anshan 243002, China.
School of Metallurgical Engineering, Anhui University of Technology, Ma'anshan 243032, China.
Int J Mol Sci. 2020 Mar 25;21(7):2274. doi: 10.3390/ijms21072274.
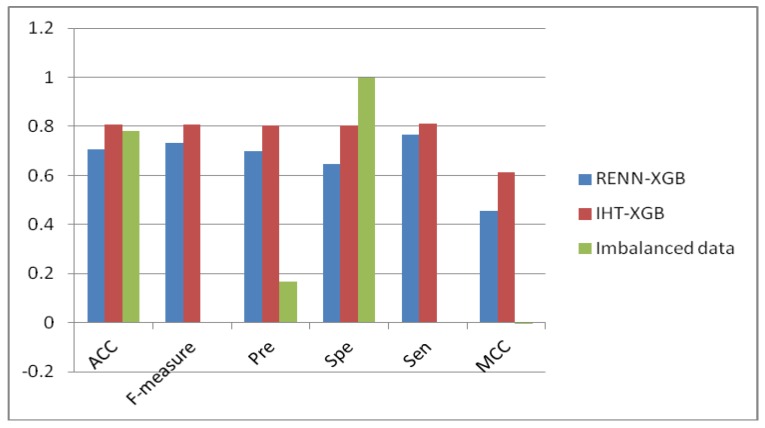
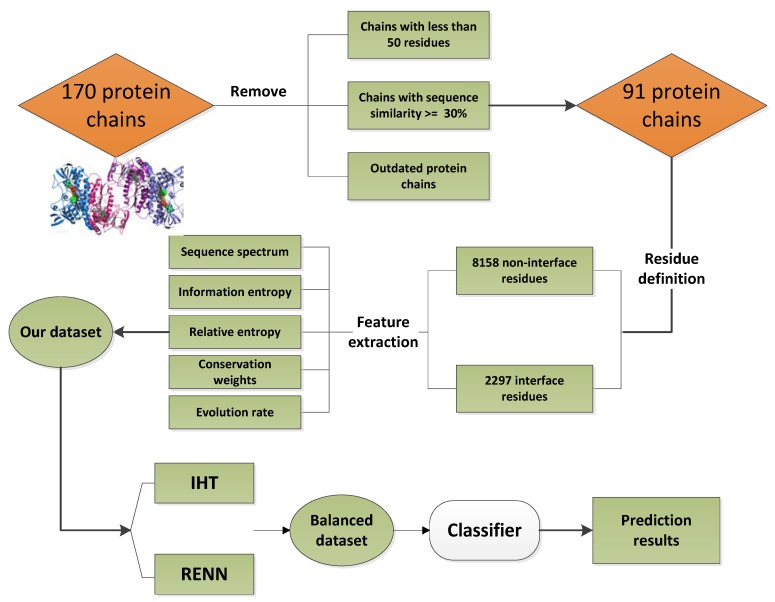
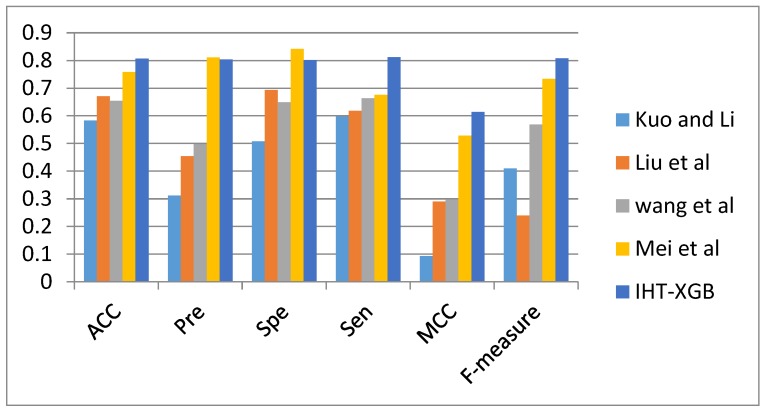
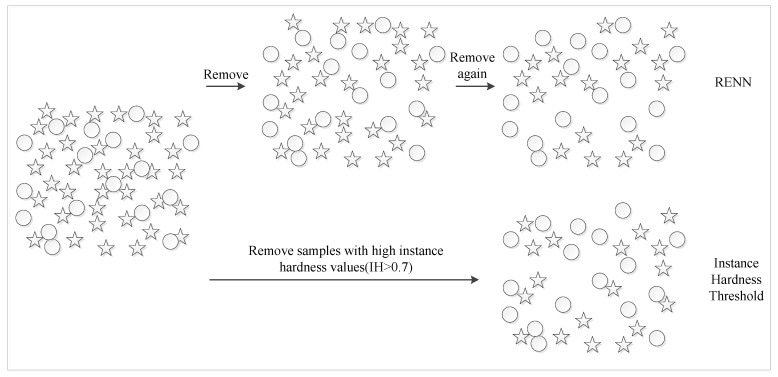
The study of protein-protein interaction is of great biological significance, and the prediction of protein-protein interaction sites can promote the understanding of cell biological activity and will be helpful for drug development. However, uneven distribution between interaction and non-interaction sites is common because only a small number of protein interactions have been confirmed by experimental techniques, which greatly affects the predictive capability of computational methods. In this work, two imbalanced data processing strategies based on XGBoost algorithm were proposed to re-balance the original dataset from inherent relationship between positive and negative samples for the prediction of protein-protein interaction sites. Herein, a feature extraction method was applied to represent the protein interaction sites based on evolutionary conservatism of proteins, and the influence of overlapping regions of positive and negative samples was considered in prediction performance. Our method showed good prediction performance, such as prediction accuracy of 0.807 and MCC of 0.614, on an original dataset with 10,455 surface residues but only 2297 interface residues. Experimental results demonstrated the effectiveness of our XGBoost-based method.
蛋白质-蛋白质相互作用的研究具有重要的生物学意义,而预测蛋白质相互作用位点可以促进对细胞生物活性的理解,并有助于药物开发。然而,由于只有少数蛋白质相互作用已经通过实验技术得到证实,因此交互和非交互位点之间的分布不均匀是很常见的,这极大地影响了计算方法的预测能力。在这项工作中,提出了两种基于 XGBoost 算法的不平衡数据处理策略,以便通过正负样本之间的固有关系重新平衡原始数据集,从而对蛋白质相互作用位点进行预测。在这里,应用了一种特征提取方法,基于蛋白质的进化保守性来表示蛋白质相互作用位点,并在预测性能中考虑了正负样本重叠区域的影响。我们的方法在一个原始数据集上表现出了良好的预测性能,例如在一个包含 10455 个表面残基但只有 2297 个界面残基的数据集上,预测准确率为 0.807,MCC 为 0.614。实验结果证明了我们基于 XGBoost 的方法的有效性。