Chen Xuan, Wang Jingyao, Peng Xiqi, Liu Kaihao, Zhang Chunduo, Zeng Xingzhen, Lai Yongqing
Guangdong and Shenzhen Key Laboratory of Male Reproductive Medicine and Genetics, Peking University Shenzhen Hospital, Institute of Urology of Shenzhen PKU-HKUST Medical Center, Shenzhen.
Shantou University Medical College, Shantou, Guangdong.
Medicine (Baltimore). 2020 Apr;99(14):e19628. doi: 10.1097/MD.0000000000019628.
Prostate cancer (PCa) is one of the leading causes of cancer-related death. In the present research, we adopted a comprehensive bioinformatics method to identify some biomarkers associated with the tumor progression and prognosis of PCa.
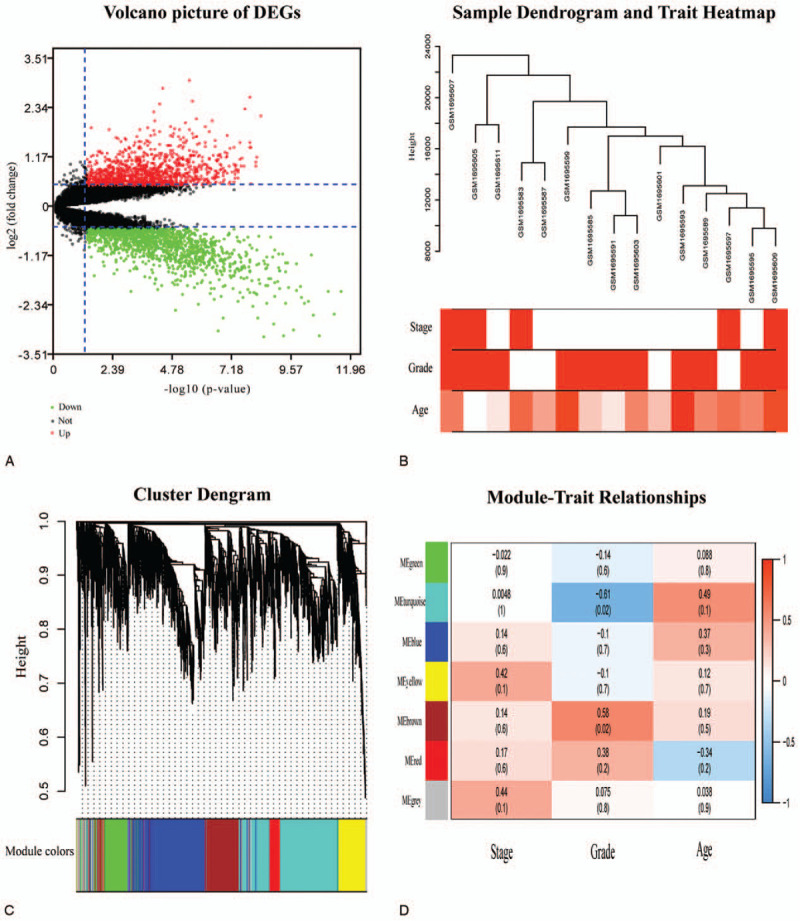
Differentially expressed genes (DEGs) analysis and weighted gene co-expression network analysis (WGCNA) were applied for exploring gene modules correlative with tumor progression and prognosis of PCa. Clinically Significant Modules were distinguished, and Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis were used to Annotation, Visualization and Integrated Discovery (DAVID). Protein-protein interaction (PPI) networks were used in selecting potential hub genes. RNA-Seq data and clinical materials of prostate cancer from The Cancer Genome Atlas (TCGA) database were used for the identification and validation of hub genes. The significance of these genes was confirmed via survival analysis and immunohistochemistry.
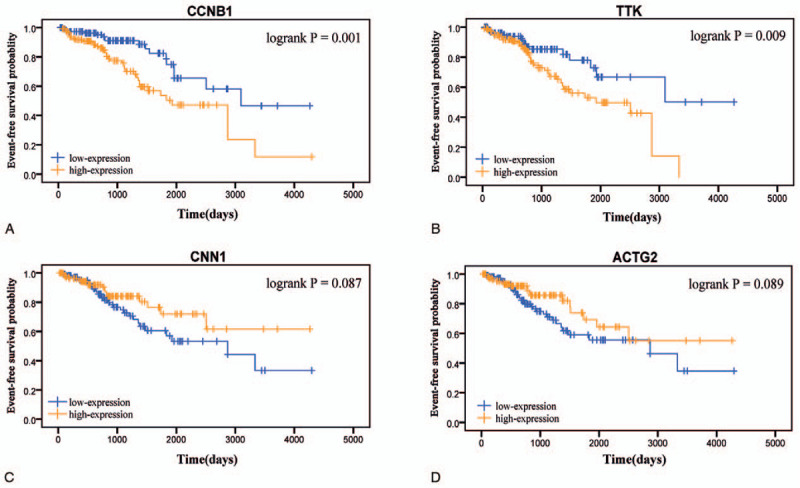
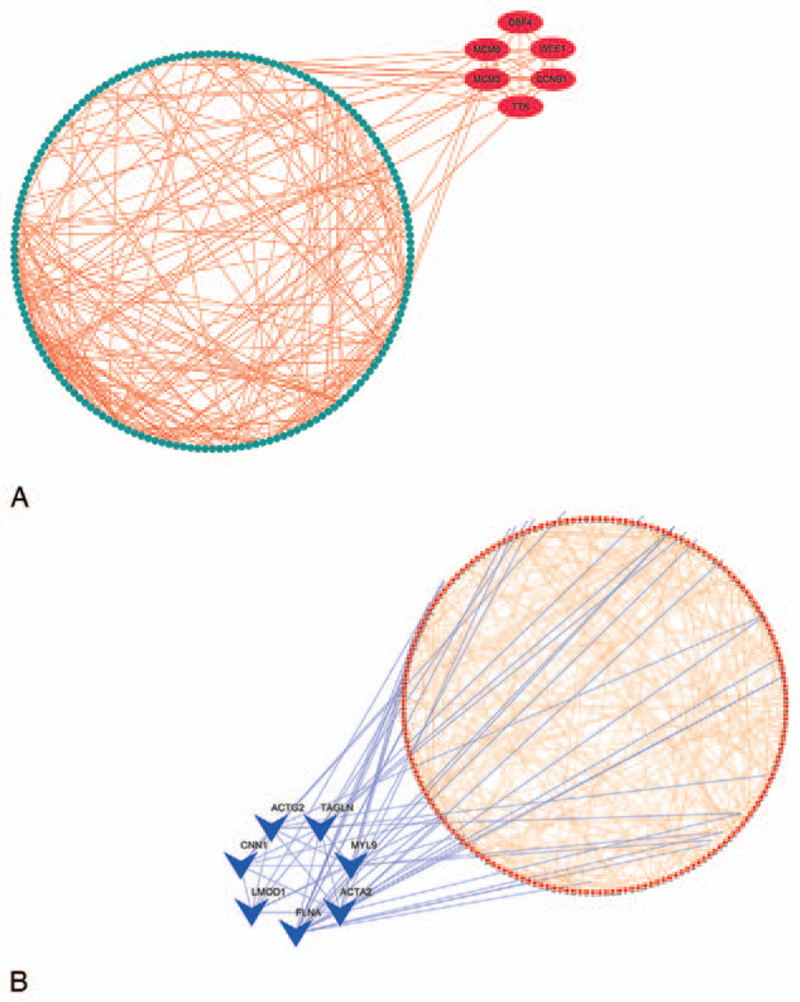
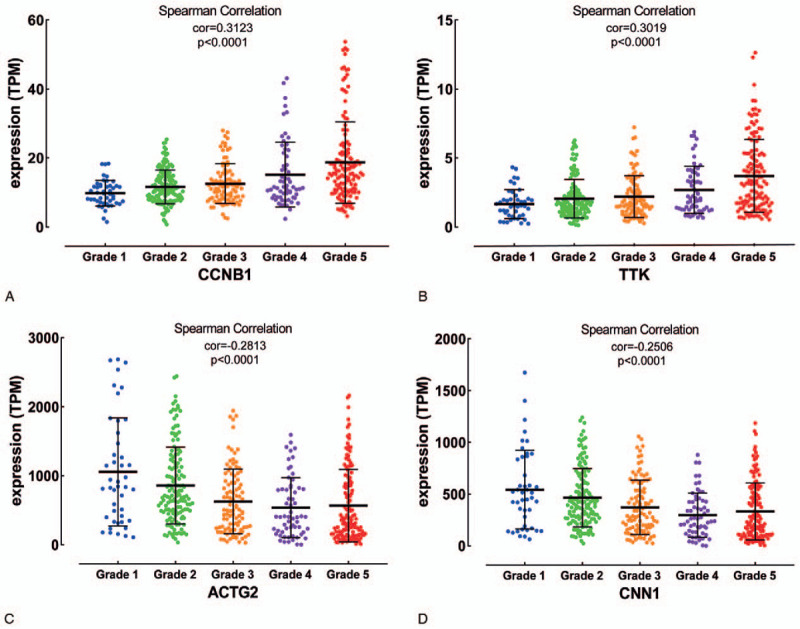
2688 DEGs were filtered. Weighted gene co-expression network was constructed, and DEGs were divided into 6 modules. Two modules were selected as hub modules which were highly associated with the tumor grades. Functional enrichment analysis was performed on genes in hub modules. Thirteen hub genes in these hub modules were identified through PPT networks. Based on TCGA data, 4 of them (CCNB1, TTK, CNN1, and ACTG2) were correlated with prognosis. The protein levels of CCNB1, TTK, and ACTG2 had a degree of differences between tumor tissues and normal tissues.
Four hub genes were identified as candidate biomarkers and potential therapeutic targets for further studies of exploring molecular mechanisms and individual therapy on PCa.
前列腺癌(PCa)是癌症相关死亡的主要原因之一。在本研究中,我们采用综合生物信息学方法来识别一些与PCa肿瘤进展和预后相关的生物标志物。
应用差异表达基因(DEG)分析和加权基因共表达网络分析(WGCNA)来探索与PCa肿瘤进展和预后相关的基因模块。区分临床显著模块,并使用基因本体(GO)和京都基因与基因组百科全书(KEGG)分析进行注释、可视化和综合发现(DAVID)。蛋白质-蛋白质相互作用(PPI)网络用于选择潜在的枢纽基因。来自癌症基因组图谱(TCGA)数据库的前列腺癌RNA测序数据和临床材料用于枢纽基因的鉴定和验证。通过生存分析和免疫组织化学证实了这些基因的意义。
筛选出2688个DEG。构建了加权基因共表达网络,DEG被分为6个模块。选择了两个模块作为枢纽模块,它们与肿瘤分级高度相关。对枢纽模块中的基因进行了功能富集分析。通过PPT网络在这些枢纽模块中鉴定出13个枢纽基因。基于TCGA数据,其中4个(CCNB1、TTK、CNN1和ACTG2)与预后相关。CCNB1、TTK和ACTG2的蛋白质水平在肿瘤组织和正常组织之间存在一定差异。
四个枢纽基因被鉴定为候选生物标志物和潜在治疗靶点,用于进一步探索PCa分子机制和个体化治疗的研究。