Department of Intensive Care Unit, The People's Hospital of Tongliang District, Chongqing, China.
Department of Respiratory and Geriatrics, Chongqing Public Health Medical Center, Chongqing, China.
Biomed Res Int. 2020 Feb 29;2020:2514230. doi: 10.1155/2020/2514230. eCollection 2020.
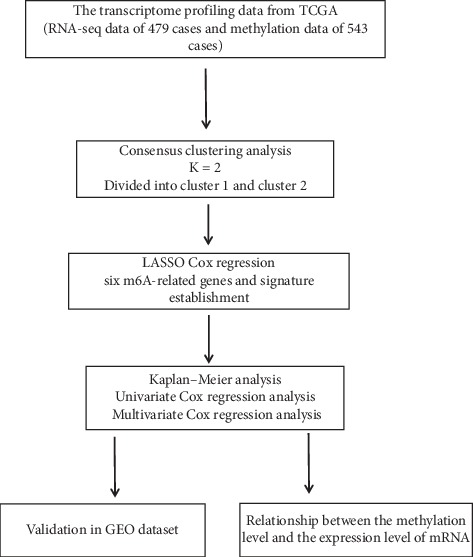
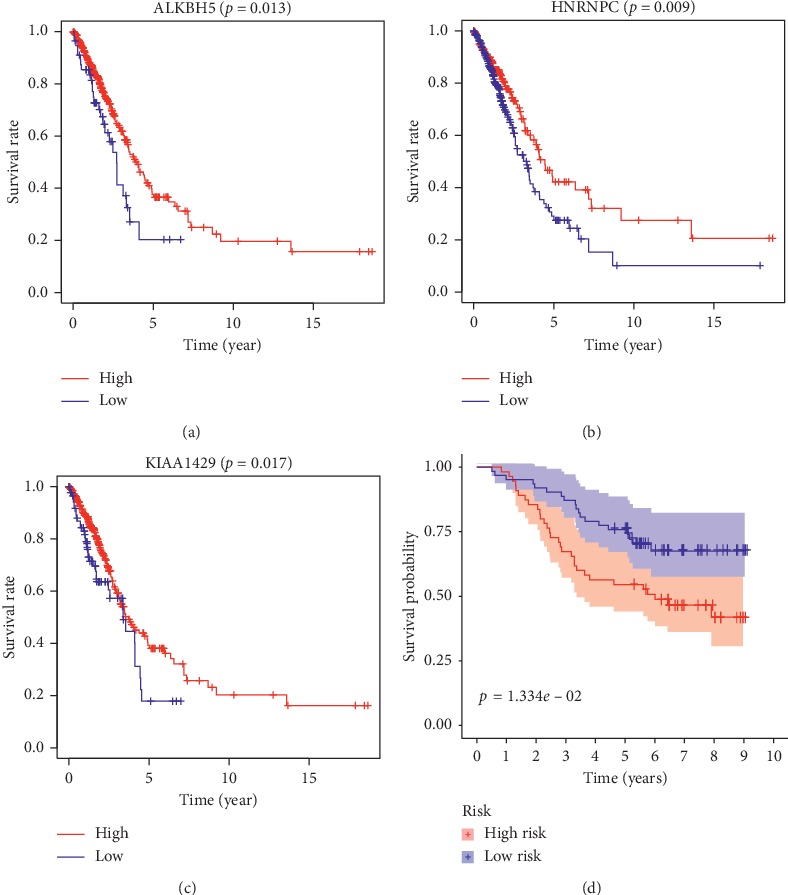
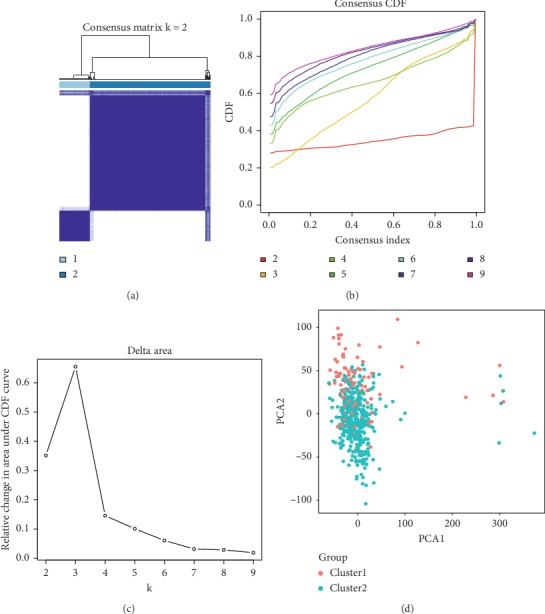
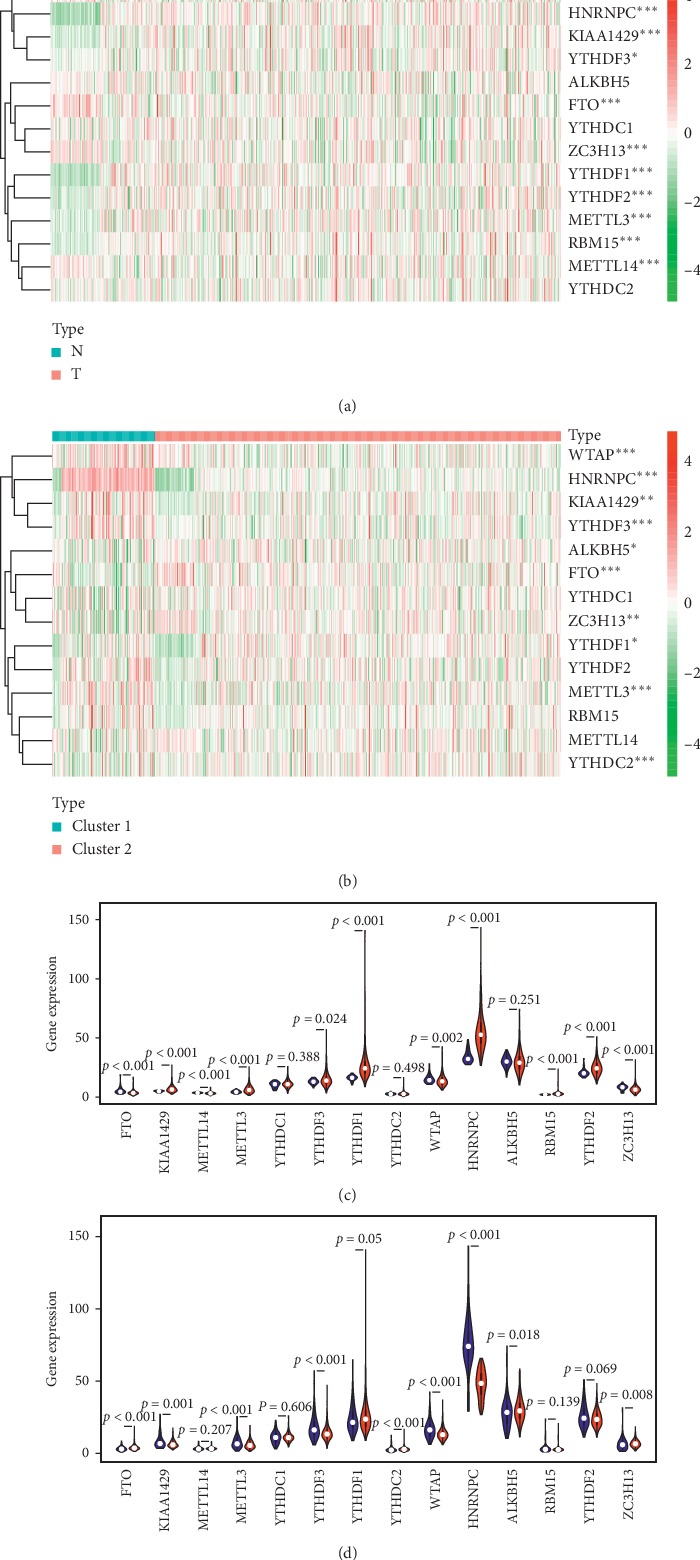
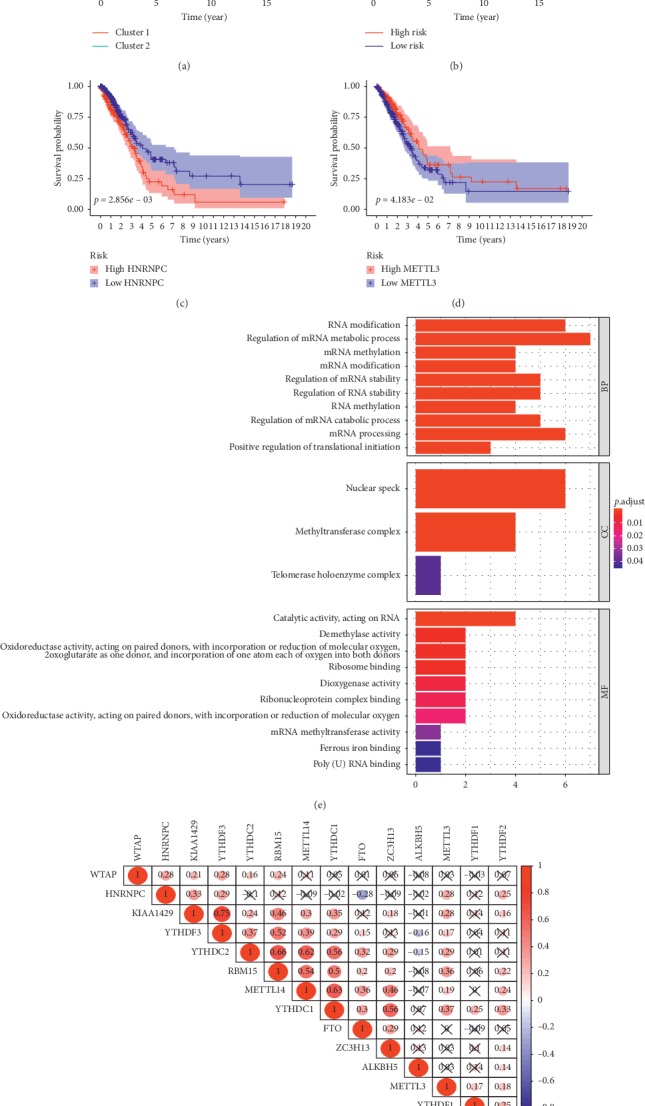
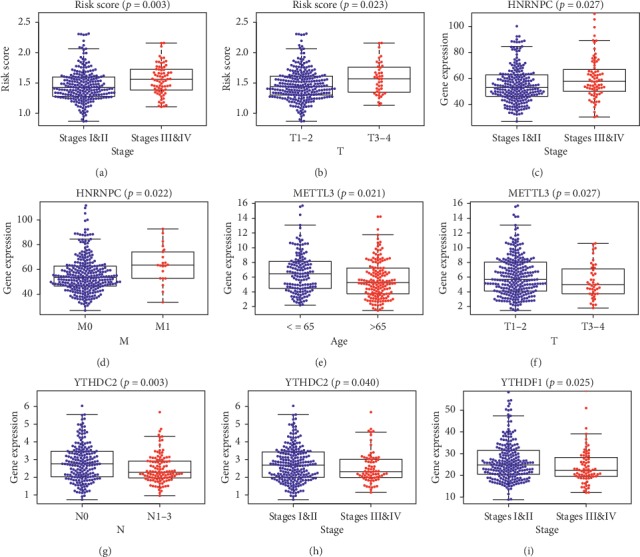
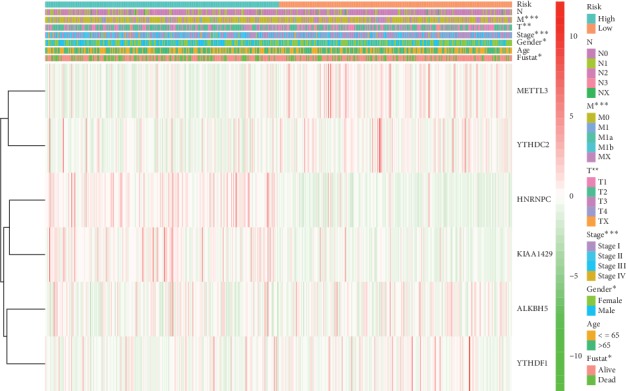
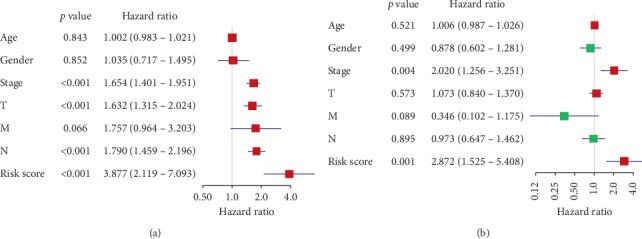
Lung cancer is the most commonly diagnosed cancer and the leading cause of cancer-related death. Among these, lung adenocarcinoma (LUAD) accounts for most cases. Due to the improvement of precision medicine based on molecular characterization, the treatment of LUAD underwent significant changes. With these changes, the prognosis of LUAD becomes diverse. N-methyladenosine (mA) is the most predominant modification in mRNAs, which has been a research hotspot in the field of oncology. Nevertheless, little has been studied to reveal the correlations between the mA-related genes and prognosis in LUAD. Thus, we conducted a comprehensive analysis of mA-related gene expressions in LUAD patients based on The Cancer Genome Atlas (TCGA) database by revealing their relationship with prognosis. Different expressions of the mA-related genes in tumor tissues and non-tumor tissues were confirmed. Furthermore, their relationship with prognosis was studied via Consensus Clustering Analysis, Principal Components Analysis (PCA), and Least Absolute Shrinkage and Selection Operator (LASSO) Regression. Based on the above analyses, a mA-based signature to predict the overall survival (OS) in LUAD was successfully established. Among the 479 cases, we found that most of the mA-related genes were differentially expressed between tumor and non-tumor tissues. Six genes, HNRNPC, METTL3, YTHDC2, KIAA1429, ALKBH5, and YTHDF1 were screened to build a risk scoring signature, which is strongly related to the clinical features pathological stages ( < 0.05), stages ( < 0.05), stages ( < 0.05), gender ( = 0.04), and survival outcome ( = 0.02). Multivariate Cox analysis indicated that risk value could be used as an independent prognostic factor, revealing that the mA-related genes signature has great predictive value. Its efficacy was also validated by data from the Gene Expression Omnibus (GEO) database.
肺癌是最常见的癌症类型,也是癌症相关死亡的主要原因。在这些癌症中,肺腺癌 (LUAD) 占大多数病例。由于基于分子特征的精准医学的进步,LUAD 的治疗方法发生了重大变化。随着这些变化,LUAD 的预后变得多样化。N6-甲基腺苷 (mA) 是 mRNA 中最主要的修饰物,它已成为肿瘤学领域的研究热点。然而,对于揭示 mA 相关基因与 LUAD 预后之间的相关性,研究甚少。因此,我们通过揭示它们与预后的关系,基于癌症基因组图谱 (TCGA) 数据库对 LUAD 患者的 mA 相关基因表达进行了全面分析。确认了肿瘤组织和非肿瘤组织中 mA 相关基因的不同表达。此外,通过共识聚类分析、主成分分析 (PCA) 和最小绝对收缩和选择算子 (LASSO) 回归研究了它们与预后的关系。基于上述分析,成功建立了一个基于 mA 的 LUAD 总生存期 (OS) 预测签名。在 479 例病例中,我们发现大多数 mA 相关基因在肿瘤组织和非肿瘤组织之间存在差异表达。筛选出 6 个基因 HNRNPC、METTL3、YTHDC2、KIAA1429、ALKBH5 和 YTHDF1 构建风险评分签名,与临床特征病理分期(<0.05)、分期(<0.05)、分期(<0.05)、性别(=0.04)和生存结果(=0.02)密切相关。多变量 Cox 分析表明,风险值可用作独立预后因素,表明 mA 相关基因签名具有很大的预测价值。它的功效也通过基因表达综合数据库 (GEO) 数据库的数据得到了验证。