Department of Gastroenterology, Faculty of Medicine, University Hospital, Slovak Medical University, Bratislava, Slovakia.
Department of Internal medicine, Hospital Poprad, Poprad, Slovakia.
BMC Gastroenterol. 2020 Apr 7;20(1):95. doi: 10.1186/s12876-020-01237-8.
Aceruloplasminaemia is a very rare autosomal recessive disorder caused by a mutation in the ceruloplasmin gene, which is clinically manifested by damage to the nervous system and retinal degeneration. This classical clinical picture can be preceded by diabetes mellitus and microcytic anaemia, which are considered to be early manifestations of aceruloplasminaemia.
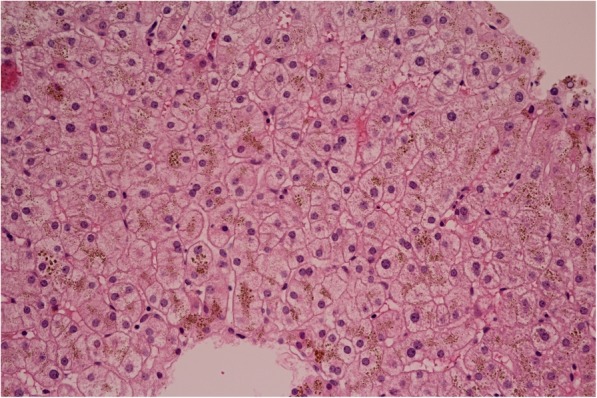
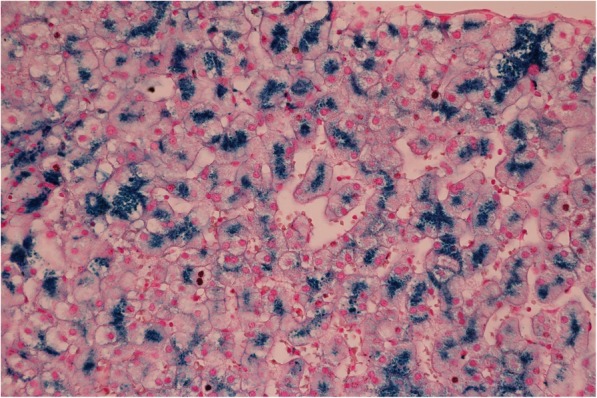
In our report, we describe the case of a patient with aceruloplasminaemia detected in an early stage (without clinical symptoms of damage to the nervous system) during the search for the cause of hepatopathy with very low values of serum ceruloplasmin. Molecular genetic examination of the CP gene for ceruloplasmin identified a new variant c.1664G > A (p.Gly555Glu) in the homozygous state, which has not been published in the literature or population frequency databases to date. Throughout the 21-month duration of chelatase treatment, the patient, who is 43 years old, continues to be without neurological and psychiatric symptomatology. We observed a decrease in the serum concentration of ferritin without a reduction in iron deposits in the brain on magnetic resonance imaging.
Currently, there is no unequivocal recommendation of an effective treatment for aceruloplasminaemia. Early diagnosis is important in the neurologically asymptomatic stage.
亚铁氧化酶缺乏症是一种非常罕见的常染色体隐性遗传病,由铜蓝蛋白基因的突变引起,其临床特征为神经系统损伤和视网膜变性。这种典型的临床表现可先于糖尿病和小细胞性贫血出现,后者被认为是亚铁氧化酶缺乏症的早期表现。
在我们的报告中,我们描述了一例在寻找肝病病因时(血清铜蓝蛋白值极低,无神经系统损伤的临床症状)早期发现的亚铁氧化酶缺乏症患者。对铜蓝蛋白 CP 基因进行分子遗传学检查,发现该患者纯合子状态存在一种新的变异 c.1664G>A(p.Gly555Glu),这一变异尚未在文献或人群频率数据库中报道过。在为期 21 个月的螯合剂治疗过程中,这位 43 岁的患者仍然没有出现神经和精神症状。我们观察到血清铁蛋白浓度降低,而磁共振成像上脑内铁沉积没有减少。
目前,对于亚铁氧化酶缺乏症尚无明确的有效治疗方法。在无症状神经阶段早期诊断很重要。