Department of Medical Oncology, Institut Roi Albert II, Cliniques universitaires Saint-Luc and Institut de Recherche Expérimentale et Clinique, UCLouvain, Brussels, Belgium.
Human Molecular Genetics, de Duve Institute, UCLouvain, Brussels, Belgium.
Breast Cancer Res. 2020 Apr 15;22(1):36. doi: 10.1186/s13058-020-01273-y.
Multigene panels are routinely used to assess for predisposing germline mutations in families at high breast cancer risk. The number of variants of unknown significance thereby identified increases with the number of sequenced genes. We aimed to determine whether tumor sequencing can help refine the analysis of germline variants based on second somatic genetic events in the same gene.
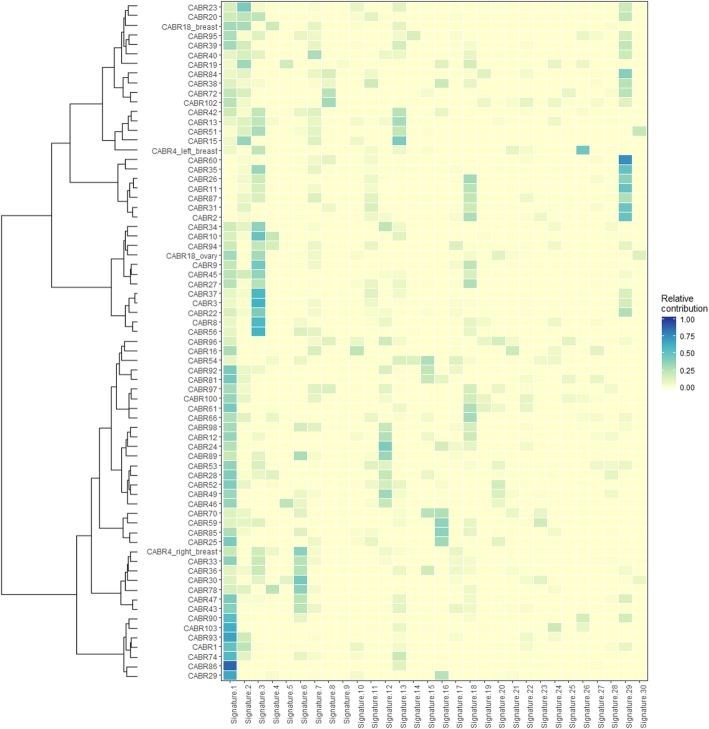
Whole-exome sequencing (WES) was performed on whole blood DNA from 70 unrelated breast cancer patients referred for genetic testing and without a BRCA1, BRCA2, TP53, or CHEK2 mutation. Rare variants were retained in a list of 735 genes. WES was performed on matched tumor DNA to identify somatic second hits (copy number alterations (CNAs) or mutations) in the same genes. Distinct methods (among which immunohistochemistry, mutational signatures, homologous recombination deficiency, and tumor mutation burden analyses) were used to further study the role of the variants in tumor development, as appropriate.
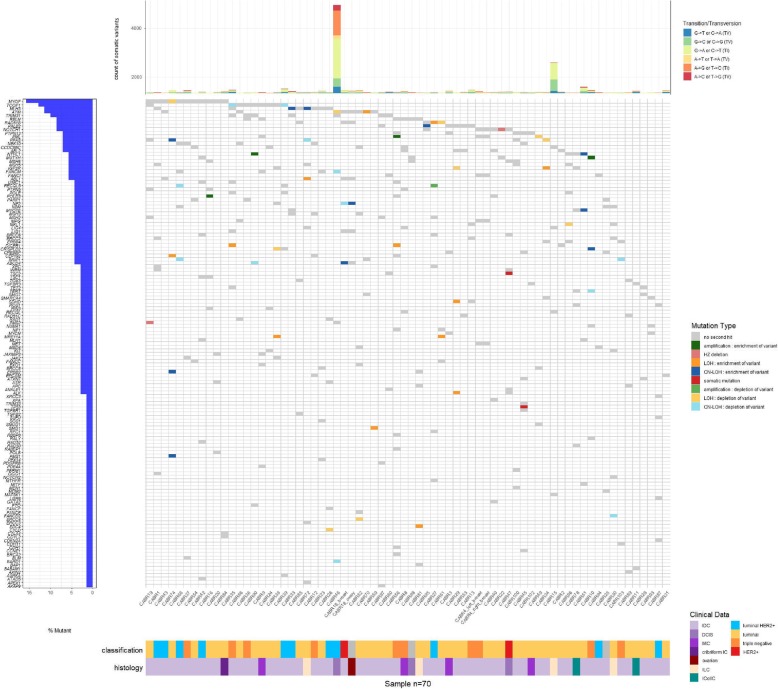
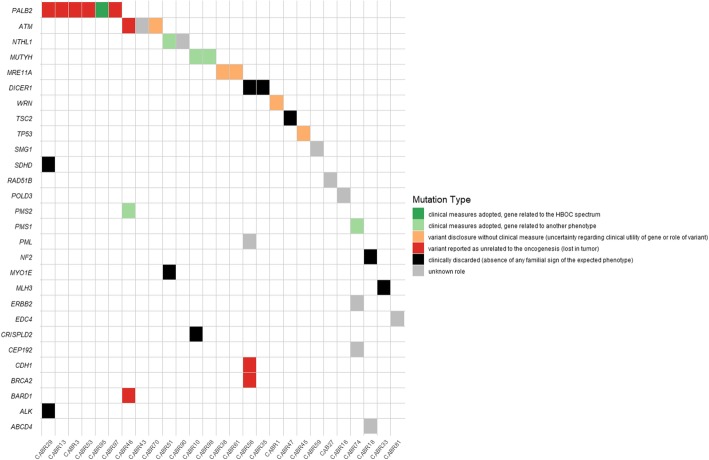
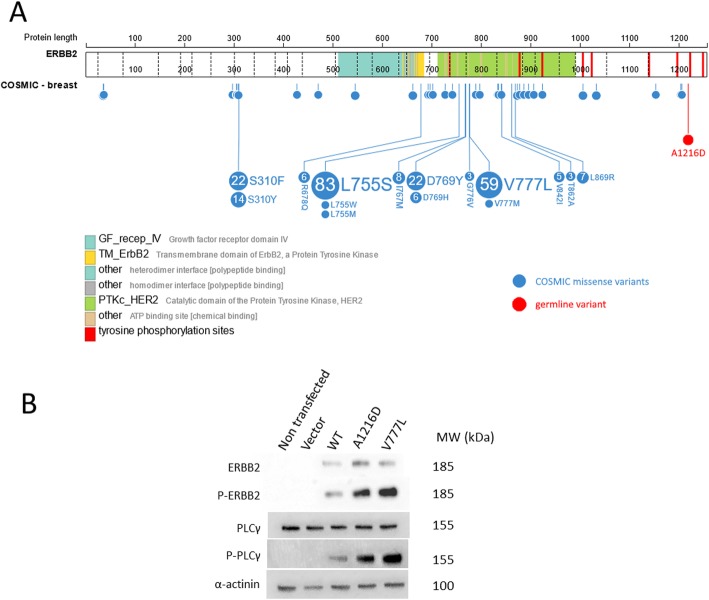
Sixty-eight patients (97%) carried at least one germline variant (4.7 ± 2.0 variants per patient). Of the 329 variants, 55 (17%) presented a second hit in paired tumor tissue. Of these, 53 were CNAs, resulting in tumor enrichment (28 variants) or depletion (25 variants) of the germline variant. Eleven patients received variant disclosure, with clinical measures for five of them. Seven variants in breast cancer-predisposing genes were considered not implicated in oncogenesis. One patient presented significant tumor enrichment of a germline variant in the oncogene ERBB2, in vitro expression of which caused downstream signaling pathway activation.
Tumor sequencing is a powerful approach to refine variant interpretation in cancer-predisposing genes in high-risk breast cancer patients. In this series, the strategy provided clinically relevant information for 11 out of 70 patients (16%), adapted to the considered gene and the familial clinical phenotype.
多基因面板常用于评估高乳腺癌风险家族中潜在的种系突变。随着测序基因数量的增加,未知意义的变异数量也会增加。我们旨在确定肿瘤测序是否可以帮助根据同一基因中的第二个体细胞遗传事件来完善种系变异的分析。
对 70 名无 BRCA1、BRCA2、TP53 或 CHEK2 突变的遗传性乳腺癌检测患者的全血 DNA 进行全外显子组测序(WES)。将罕见变异保留在 735 个基因列表中。对匹配的肿瘤 DNA 进行 WES,以识别同一基因中的体细胞第二击(拷贝数改变(CNAs)或突变)。使用不同的方法(包括免疫组织化学、突变特征、同源重组缺陷和肿瘤突变负担分析),根据需要进一步研究变异在肿瘤发生中的作用。
68 名患者(97%)携带至少一种种系变异(每名患者 4.7±2.0 个变异)。在 329 个变异中,55 个(17%)在配对肿瘤组织中存在第二击。其中 53 个是 CNA,导致种系变异的肿瘤富集(28 个变异)或耗竭(25 个变异)。11 名患者接受了变异披露,其中 5 名患者进行了临床评估。在易患乳腺癌的基因中,有 7 个变异被认为与致癌作用无关。一名患者的致癌基因 ERBB2 中存在种系变异的显著肿瘤富集,体外表达导致下游信号通路激活。
肿瘤测序是一种强大的方法,可以在高风险乳腺癌患者的癌症易感基因中完善变异解释。在本系列中,该策略为 70 名患者中的 11 名(16%)提供了临床相关信息,该策略适应所考虑的基因和家族临床表型。