Medical Genomics Group, QIMR Berghofer Medical Research Institute, Brisbane, QLD.
Faculty of Medicine, Centre for Clinical Research, The University of Queensland, Brisbane, QLD.
Ann Oncol. 2019 Jul 1;30(7):1071-1079. doi: 10.1093/annonc/mdz132.
Whole-genome sequencing (WGS) is a powerful method for revealing the diversity and complexity of the somatic mutation burden of tumours. Here, we investigated the utility of tumour and matched germline WGS for understanding aetiology and treatment opportunities for high-risk individuals with familial breast cancer.
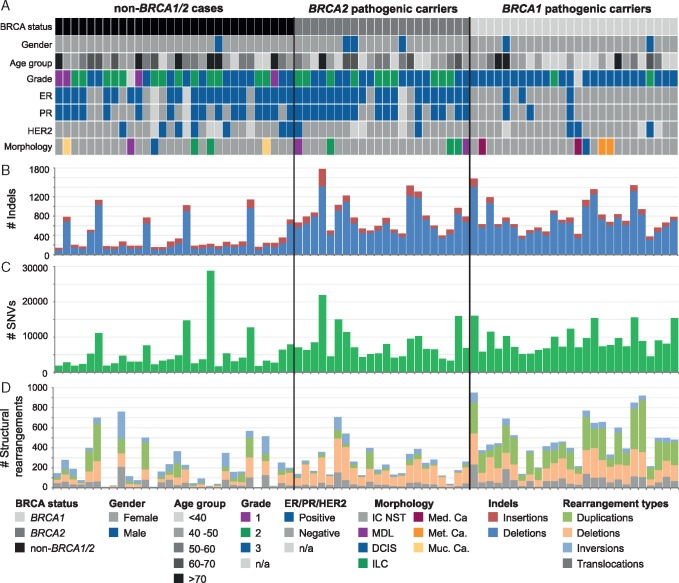
We carried out WGS on 78 paired germline and tumour DNA samples from individuals carrying pathogenic variants in BRCA1 (n = 26) or BRCA2 (n = 22) or from non-carriers (non-BRCA1/2; n = 30).
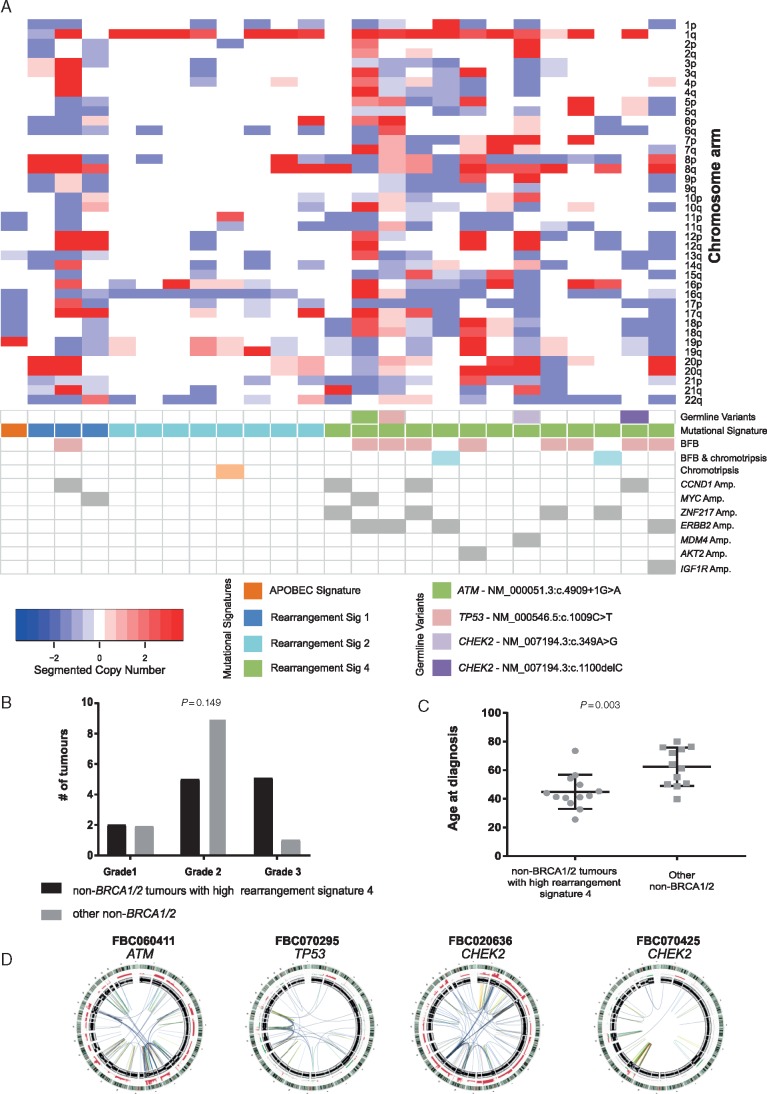
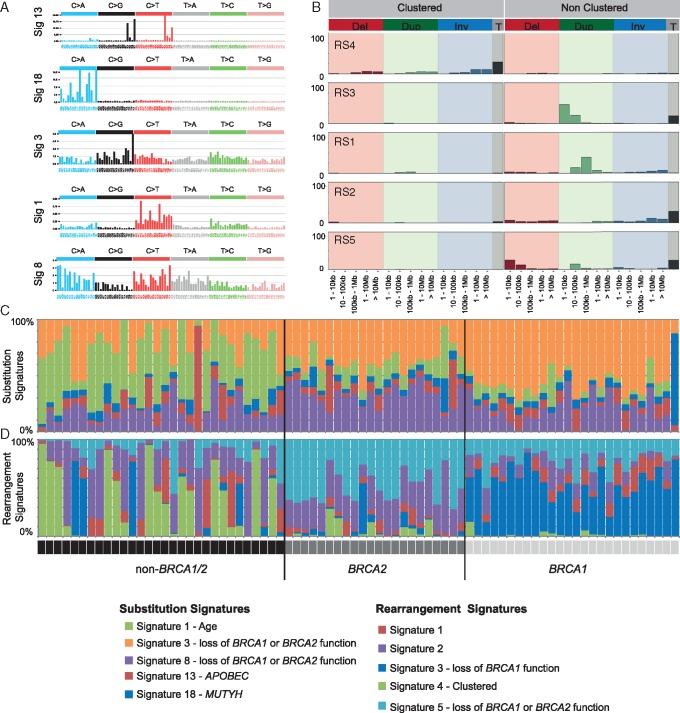
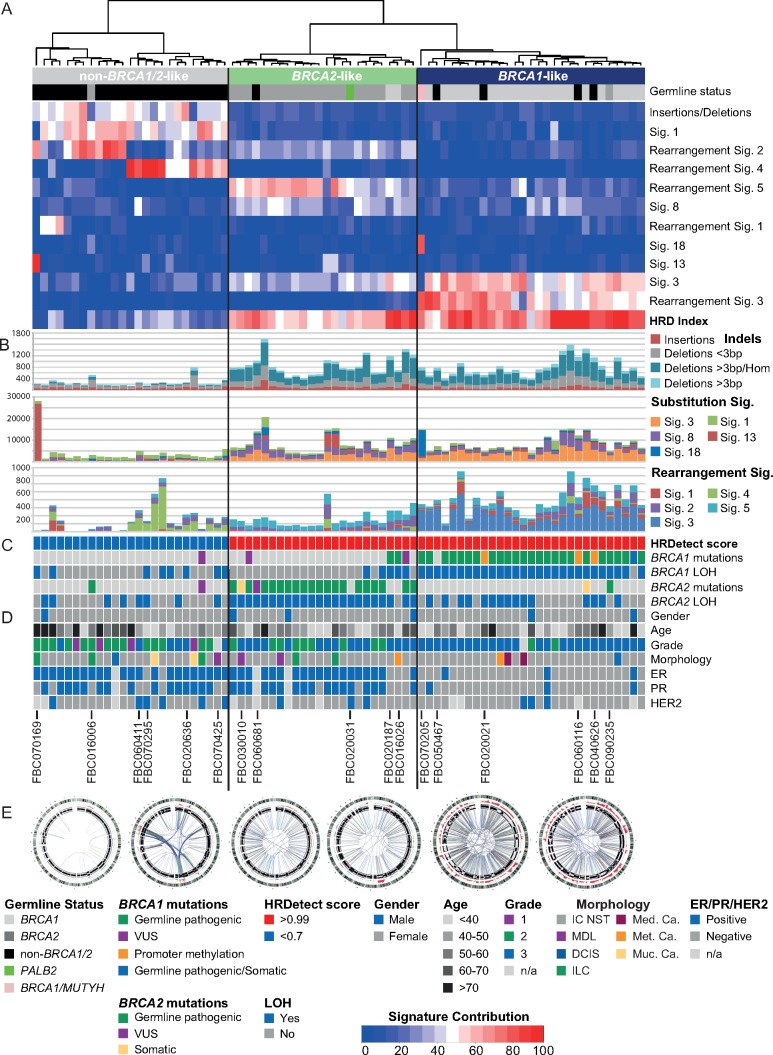
Matched germline/tumour WGS and somatic mutational signature analysis revealed patients with unreported, dual pathogenic germline variants in cancer risk genes (BRCA1/BRCA2; BRCA1/MUTYH). The strategy identified that 100% of tumours from BRCA1 carriers and 91% of tumours from BRCA2 carriers exhibited biallelic inactivation of the respective gene, together with somatic mutational signatures suggestive of a functional deficiency in homologous recombination. A set of non-BRCA1/2 tumours also had somatic signatures indicative of BRCA-deficiency, including tumours with BRCA1 promoter methylation, and tumours from carriers of a PALB2 pathogenic germline variant and a BRCA2 variant of uncertain significance. A subset of 13 non-BRCA1/2 tumours from early onset cases were BRCA-proficient, yet displayed complex clustered structural rearrangements associated with the amplification of oncogenes and pathogenic germline variants in TP53, ATM and CHEK2.
Our study highlights the role that WGS of matched germline/tumour DNA and the somatic mutational signatures can play in the discovery of pathogenic germline variants and for providing supporting evidence for variant pathogenicity. WGS-derived signatures were more robust than germline status and other genomic predictors of homologous recombination deficiency, thus impacting the selection of platinum-based or PARP inhibitor therapy. In this first examination of non-BRCA1/2 tumours by WGS, we illustrate the considerable heterogeneity of these tumour genomes and highlight that complex genomic rearrangements may drive tumourigenesis in a subset of cases.
全基因组测序(WGS)是一种强大的方法,可以揭示肿瘤体细胞突变负担的多样性和复杂性。在这里,我们研究了肿瘤和匹配的种系 WGS 在了解携带家族性乳腺癌高危个体的病因和治疗机会方面的效用。
我们对携带 BRCA1(n=26)或 BRCA2 种系变异(n=22)的个体或非携带者(非 BRCA1/2;n=30)的 78 对种系和肿瘤 DNA 样本进行了 WGS。
匹配的种系/肿瘤 WGS 和体细胞突变特征分析揭示了具有未报告的癌症风险基因(BRCA1/BRCA2;BRCA1/MUTYH)双种系致病性变异的患者。该策略确定 100%的 BRCA1 携带者肿瘤和 91%的 BRCA2 携带者肿瘤均表现出相应基因的双等位基因失活,同时具有同源重组功能缺陷的体细胞突变特征。一组非 BRCA1/2 肿瘤也具有 BRCA 缺陷的体细胞特征,包括 BRCA1 启动子甲基化的肿瘤,以及携带 PALB2 种系致病性变异和 BRCA2 意义未明变异的肿瘤。一组来自早发性病例的 13 例非 BRCA1/2 肿瘤为 BRCA 阳性,但显示出与癌基因扩增和 TP53、ATM 和 CHEK2 中的致病性种系变异相关的复杂聚类结构重排。
我们的研究强调了匹配的种系/肿瘤 DNA 的 WGS 和体细胞突变特征在发现致病性种系变异和为变异致病性提供支持证据方面的作用。WGS 衍生的特征比种系状态和同源重组缺陷的其他基因组预测更稳健,因此影响了铂类或 PARP 抑制剂治疗的选择。在首次对非 BRCA1/2 肿瘤进行 WGS 检查中,我们说明了这些肿瘤基因组的相当大的异质性,并强调复杂的基因组重排可能在一部分病例中驱动肿瘤发生。