Beach Rianne, Abitbol Julia M, Allman Brian L, Esseltine Jessica L, Shao Qing, Laird Dale W
Department of Anatomy and Cell Biology, Schulich School of Medicine & Dentistry, University of Western Ontario, London, ON, Canada.
Division of BioMedical Sciences, Faculty of Medicine, Memorial University of Newfoundland, St. John's, NL, Canada.
Front Cell Dev Biol. 2020 Apr 2;8:215. doi: 10.3389/fcell.2020.00215. eCollection 2020.
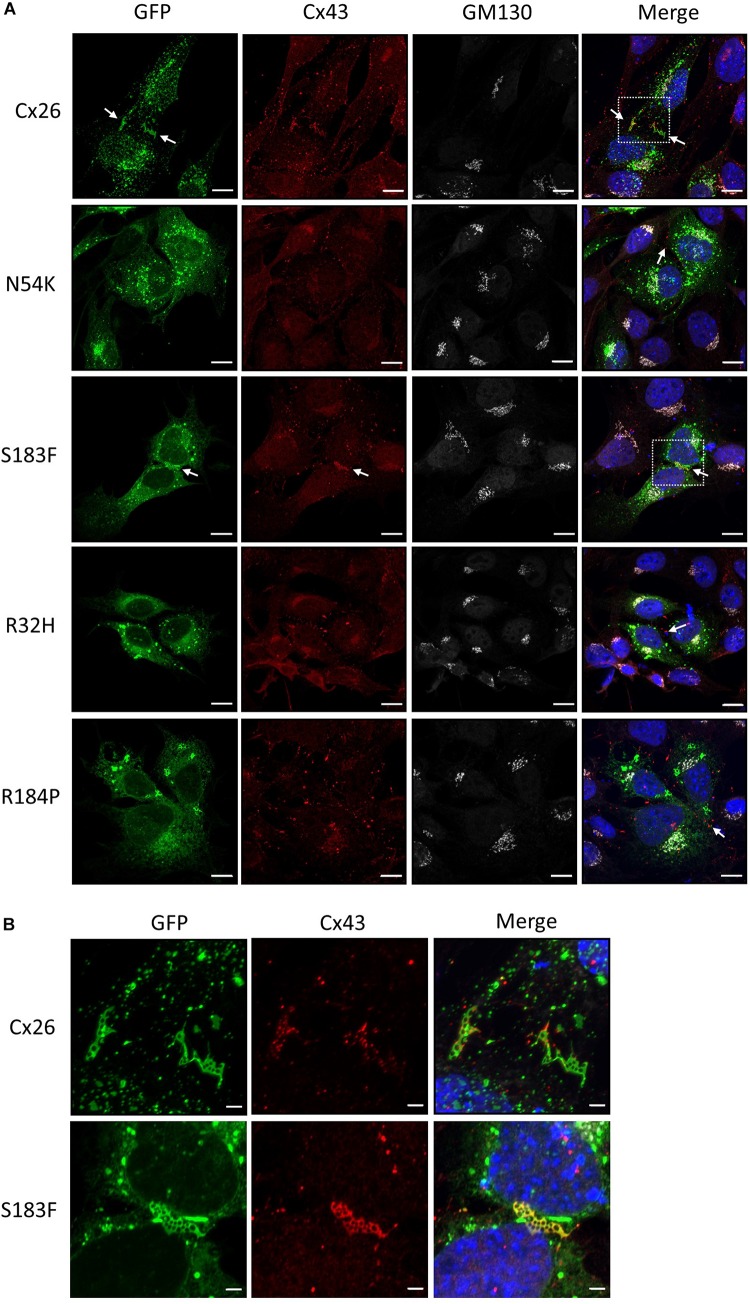
gene (that encodes Cx26) mutations are causal of hearing loss highlighting the importance of Cx26-based channel signaling amongst the supporting cells in the organ of Corti. While the majority of these mutations are inherited in an autosomal recessive manner, others are inherited in an autosomal dominant manner and lead to syndromic hearing loss as well as skin diseases. To assess if common or divergent mechanisms are at the root of -linked hearing loss, we expressed several mutants in cochlear-relevant HEI-OC1 cells derived from the developing organ of Corti. Since supporting cells of the mature mammalian organ of Corti have negligible Cx43, but HEI-OC1 cells are rich in Cx43, we first used CRISPR-Cas9 to ablate endogenous Cx43, thus establishing a connexin-deficient platform for controlled reintroduction of hearing-relevant connexins and Cx26 mutants. We found three distinct outcomes and cellular phenotypes when hearing loss-linked Cx26 mutants were expressed in cochlear-relevant cells. The dominant syndromic Cx26 mutant N54K had trafficking defects and did not fully prevent wild-type Cx26 gap junction plaque formation but surprisingly formed gap junctions when co-expressed with Cx30. In contrast, the dominant syndromic S183F mutant formed gap junctions incapable of transferring dye and, as expected, co-localized in the same gap junctions as wild-type Cx26 and Cx30, but also gained the capacity to intermix with Cx43 within gap junctions. Both recessive non-syndromic Cx26 mutants (R32H and R184P) were retained in intracellular vesicles including early endosomes and did not co-localize with Cx30. As might be predicted, none of the Cx26 mutants prevented Cx43 gap junction plaque formation in Cx43-rich HEI-OC1 cells while Cx43-ablation had little effect on the expression of reference genes linked to auditory cell differentiation. We conclude from our studies in cochlear-relevant cells that the selected Cx26 mutants likely evoke hearing loss via three unique connexin defects that are independent of Cx43 status.
编码Cx26的基因突变是听力损失的病因,这突出了基于Cx26的通道信号在柯蒂氏器支持细胞中的重要性。虽然这些突变大多以常染色体隐性方式遗传,但其他突变以常染色体显性方式遗传,并导致综合征性听力损失以及皮肤病。为了评估常见或不同的机制是否是X连锁听力损失的根源,我们在源自发育中的柯蒂氏器的与耳蜗相关的HEI-OC1细胞中表达了几种突变体。由于成熟哺乳动物柯蒂氏器的支持细胞中Cx43含量可忽略不计,但HEI-OC1细胞富含Cx43,我们首先使用CRISPR-Cas9消除内源性Cx43,从而建立一个连接蛋白缺陷平台,用于可控地重新引入与听力相关的连接蛋白和Cx26突变体。当在与耳蜗相关的细胞中表达与听力损失相关的Cx26突变体时,我们发现了三种不同的结果和细胞表型。显性综合征性Cx26突变体N54K存在转运缺陷,不能完全阻止野生型Cx26间隙连接斑的形成,但令人惊讶的是,当与Cx30共表达时会形成间隙连接。相比之下,显性综合征性S183F突变体形成了不能转移染料的间隙连接,并且如预期的那样,与野生型Cx26和Cx30共定位在相同的间隙连接中,但也获得了在间隙连接内与Cx43混合的能力。两种隐性非综合征性Cx26突变体(R32H和R184P)保留在细胞内囊泡中,包括早期内体,并且不与Cx30共定位。正如可能预测的那样,在富含Cx43的HEI-OC1细胞中,没有一种Cx26突变体阻止Cx43间隙连接斑的形成,而Cx43的消除对与听觉细胞分化相关的参考基因的表达几乎没有影响。我们从对与耳蜗相关细胞的研究中得出结论,所选的Cx26突变体可能通过三种独立于Cx43状态的独特连接蛋白缺陷引起听力损失。