Olaiya Oluwatobi R, Oyesile Doyinsola, Stone Nicholas, Mbuagbaw Lawrence, McRae Mark H
Michael G. DeGroote School of Medicine, McMaster University, Hamilton, Ontario, Canada.
University of Western Ontario Faculty of Health Science, London, Ontario, Canada.
Plast Reconstr Surg Glob Open. 2020 Feb 19;8(2):e2621. doi: 10.1097/GOX.0000000000002621. eCollection 2020 Feb.
In the United States, high-risk medical devices must be cleared through the premarket approval (PMA) pathway, which requires clinical evidence ensuring safety and efficacy. Approved devices can be modified and reintroduced to market without additional study through the PMA supplemental review track. This study characterizes the changes of high-risk plastic surgery devices once they undergo initial clearance.
A retrospective, cross-sectional analysis of the Food and Drug Administration (FDA) PMA database. The following data were extracted from the PMA database (January 1, 1980 to December 31, 2018): initial clearance date, device type, the number and type of supplement, supplement reason, and product withdrawal date. Data from the FDA medical device recall database were also extracted and reported. The median number of device modifications and median lifetime of device-years were calculated.
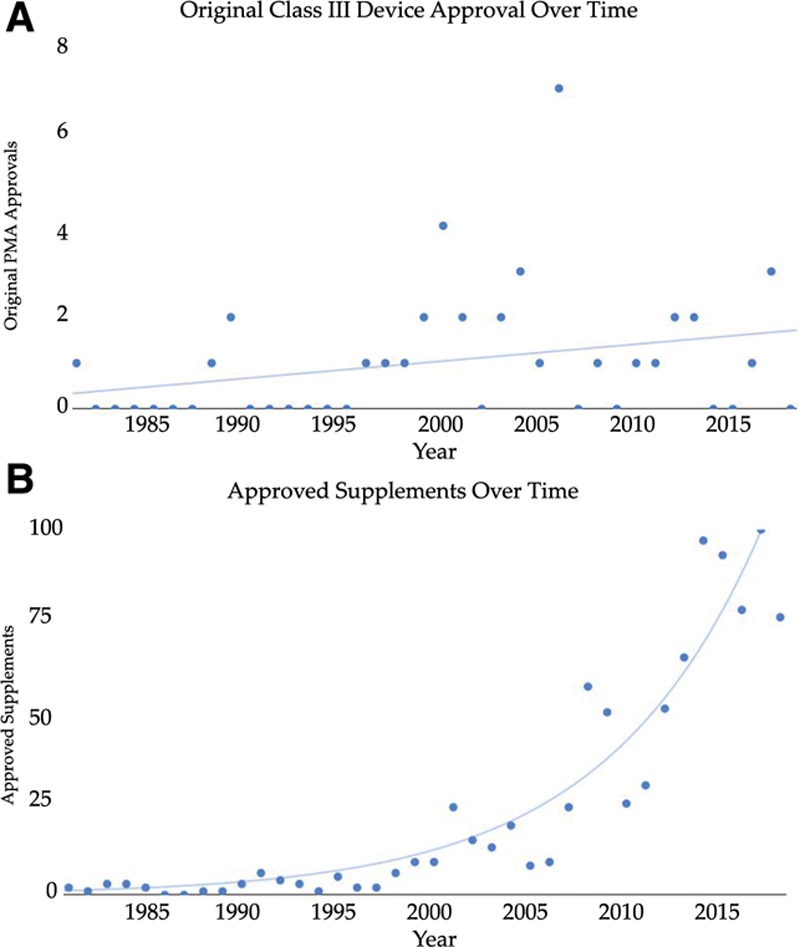
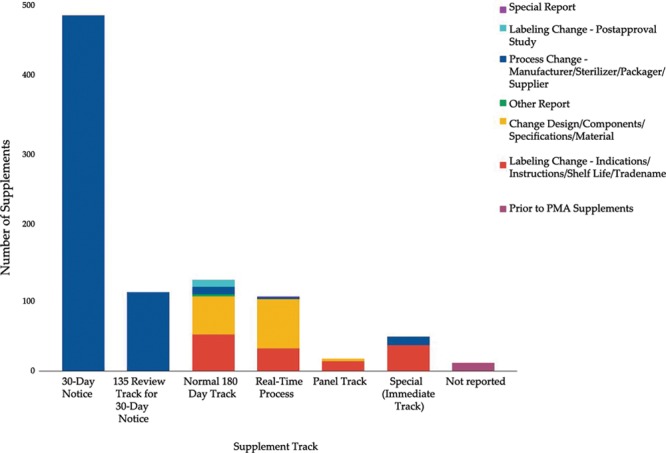
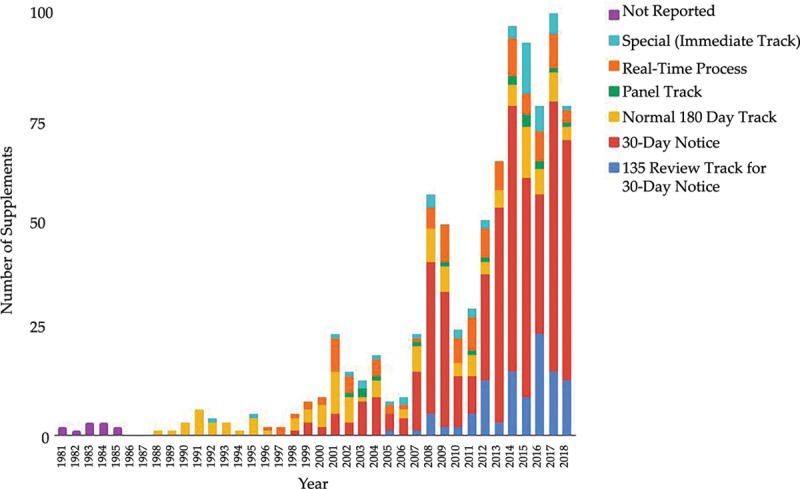
There have been 39 original plastic surgery devices approved by the FDA. There was no significant change with respect to initial clearance dates for original devices over time (r = 0.28; = 0.084). PMA supplement usage has significantly increased with time (r = 0.9174, = 0.000). Overall, approved plastic surgery devices have undergone a median of 11 changes (IQR, 3-35). Breast implant devices collectively underwent the most modifications with a median of 28 modifications per device (IQR, 20.25-33.25).
Over the past 2 decades, plastic surgery device manufacturers have significantly increased the use of supplement track review. High-risk plastic surgery devices may undergo frequent minor changes without clinical evidence to support the safety and efficacy of modified versions.
在美国,高风险医疗器械必须通过上市前批准(PMA)途径获得批准,这需要临床证据来确保安全性和有效性。已批准的器械可以通过PMA补充审查途径进行修改并重新推向市场,而无需进行额外研究。本研究描述了高风险整形手术器械在首次获得批准后的变化情况。
对美国食品药品监督管理局(FDA)的PMA数据库进行回顾性横断面分析。从PMA数据库(1980年1月1日至2018年12月31日)中提取以下数据:首次批准日期、器械类型、补充申请的数量和类型、补充申请原因以及产品撤回日期。还提取并报告了FDA医疗器械召回数据库中的数据。计算器械修改的中位数数量和器械使用年限的中位数。
FDA已批准39种原始整形手术器械。随着时间的推移,原始器械的首次批准日期没有显著变化(r = 0.28;P = 0.084)。PMA补充申请的使用随时间显著增加(r = 0.9174,P = 0.000)。总体而言,已批准的整形手术器械平均经历了11次变更(四分位距,3 - 35)。乳房植入器械的变更最为频繁,每个器械平均变更28次(四分位距,20.25 - 33.25)。
在过去20年中,整形手术器械制造商显著增加了补充审查途径的使用。高风险整形手术器械可能会频繁进行微小变更,而没有临床证据支持修改后版本的安全性和有效性。