Zhang Wei, Zeng Yifu, Wang Lei, Liu Yue, Cheng Yi-Nan
College of Computer Engineering and Applied Mathematics, Changsha University, Changsha, China.
Hunan Province Key Laboratory of Industrial Internet Technology and Security, Changsha University, Changsha, China.
Front Bioeng Biotechnol. 2020 Apr 7;8:271. doi: 10.3389/fbioe.2020.00271. eCollection 2020.
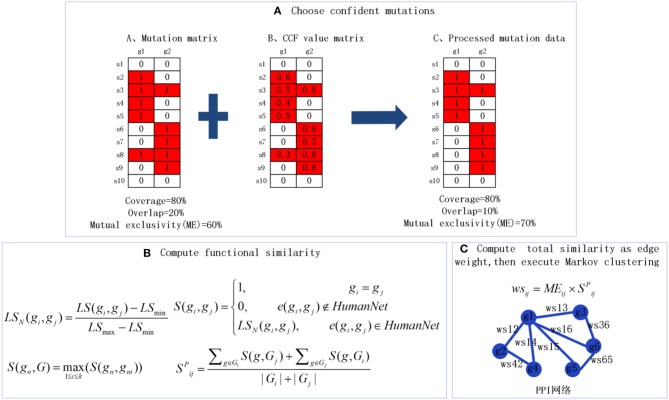
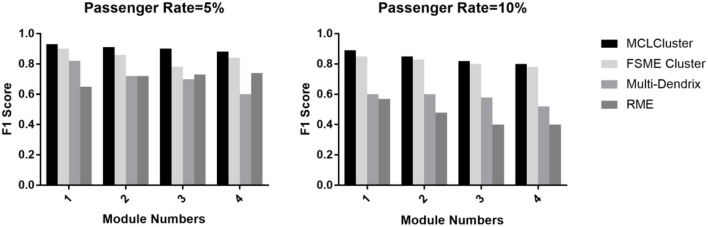
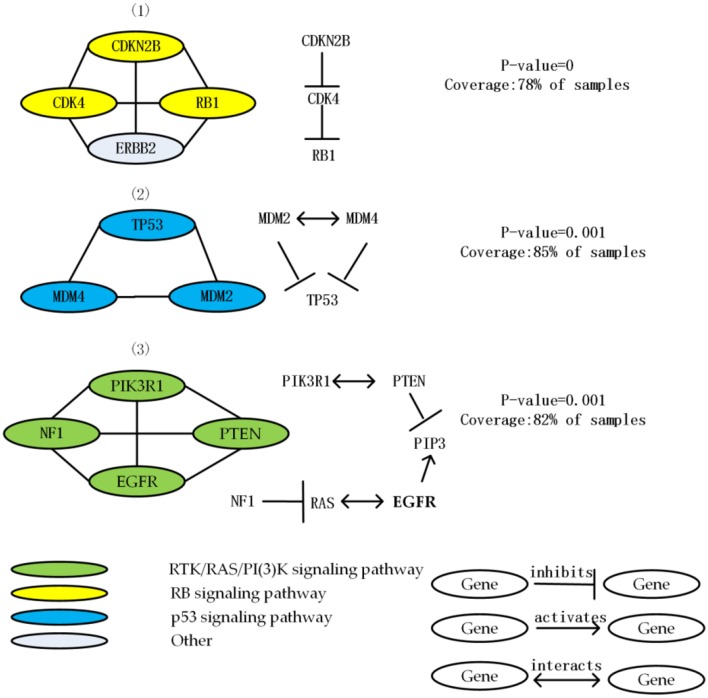
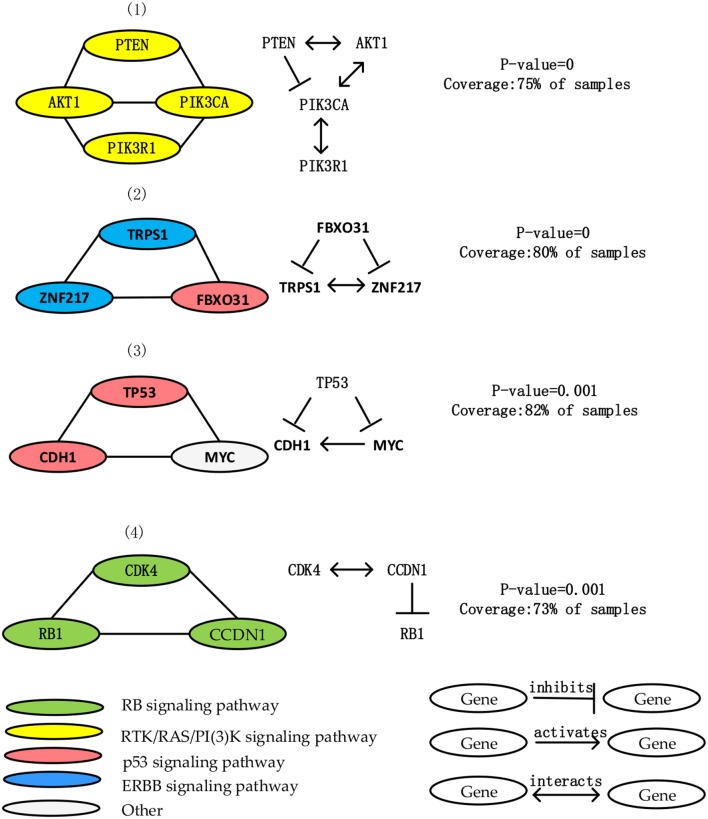
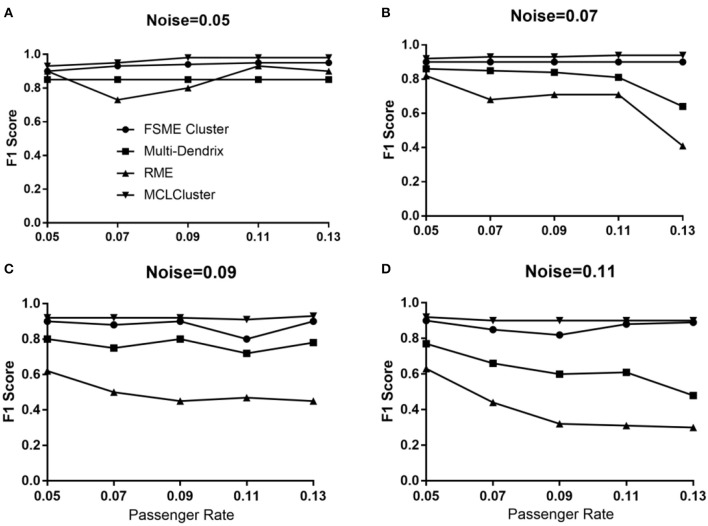
Identifying the molecular modules that drive cancer progression can greatly deepen the understanding of cancer mechanisms and provide useful information for targeted therapies. Most methods currently addressing this issue primarily use mutual exclusivity without making full use of the extra layer of module property. In this paper, we propose MCLCluster to identity cancer driver modules, which use somatic mutation data, Cancer Cell Fraction (CCF) data, gene functional interaction network and protein-protein interaction (PPI) network to derive the module property on mutual exclusivity, connectivity in PPI network and functionally similarity of genes. We have taken three effective measures to ensure the effectiveness of our algorithm. First, we use CCF data to choose stronger signals and more confident mutations. Second, the weighted gene functional interaction network is used to quantify the gene functional similarity in PPI. The third, graph clustering method based on Markov is exploited to extract the candidate module. MCLCluster is tested in the two TCGA datasets (GBM and BRCA), and identifies several well-known oncogenes driver modules and some modules with functionally associated driver genes. Besides, we compare it with Multi-Dendrix, FSME Cluster and RME in simulated dataset with background noise and passenger rate, MCLCluster outperforming all of these methods.
识别驱动癌症进展的分子模块能够极大地加深对癌症机制的理解,并为靶向治疗提供有用信息。目前解决这一问题的大多数方法主要使用互斥性,而没有充分利用模块属性的额外层面。在本文中,我们提出了MCLCluster来识别癌症驱动模块,该方法利用体细胞突变数据、癌细胞分数(CCF)数据、基因功能相互作用网络和蛋白质-蛋白质相互作用(PPI)网络来推导互斥性、PPI网络中的连通性以及基因功能相似性方面的模块属性。我们采取了三项有效措施来确保算法的有效性。首先,我们使用CCF数据来选择更强的信号和更可靠的突变。其次,加权基因功能相互作用网络用于量化PPI中基因的功能相似性。第三,利用基于马尔可夫的图聚类方法来提取候选模块。MCLCluster在两个TCGA数据集(胶质母细胞瘤和乳腺癌)中进行了测试,并识别出了几个知名的癌基因驱动模块以及一些具有功能相关驱动基因的模块。此外,我们在具有背景噪声和乘客率的模拟数据集中将其与Multi-Dendrix、FSME Cluster和RME进行了比较,MCLCluster的性能优于所有这些方法。