Department of Applied Mathematics, Computer Science & Statistics, Ghent University, Belgium; VIB-UGent Center for Medical Biotechnology, VIB, Ghent, Belgium; Department of Biomolecular Medicine, Ghent University, Ghent, Belgium; Bioinformatics Institute Ghent, Ghent University, Ghent, Belgium.
VIB-UGent Center for Medical Biotechnology, VIB, Ghent, Belgium; Department of Biomolecular Medicine, Ghent University, Ghent, Belgium; Bioinformatics Institute Ghent, Ghent University, Ghent, Belgium.
Mol Cell Proteomics. 2020 Jul;19(7):1209-1219. doi: 10.1074/mcp.RA119.001624. Epub 2020 Apr 22.
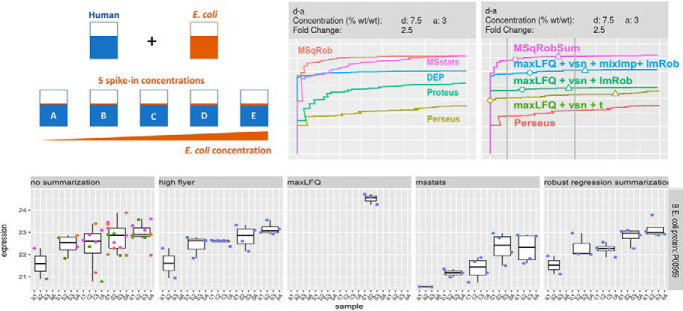
Label-Free Quantitative mass spectrometry based workflows for differential expression (DE) analysis of proteins impose important challenges on the data analysis because of peptide-specific effects and context dependent missingness of peptide intensities. Peptide-based workflows, like MSqRob, test for DE directly from peptide intensities and outperform summarization methods which first aggregate MS1 peptide intensities to protein intensities before DE analysis. However, these methods are computationally expensive, often hard to understand for the non-specialized end-user, and do not provide protein summaries, which are important for visualization or downstream processing. In this work, we therefore evaluate state-of-the-art summarization strategies using a benchmark spike-in dataset and discuss why and when these fail compared with the state-of-the-art peptide based model, MSqRob. Based on this evaluation, we propose a novel summarization strategy, MSqRobSum, which estimates MSqRob's model parameters in a two-stage procedure circumventing the drawbacks of peptide-based workflows. MSqRobSum maintains MSqRob's superior performance, while providing useful protein expression summaries for plotting and downstream analysis. Summarizing peptide to protein intensities considerably reduces the computational complexity, the memory footprint and the model complexity, and makes it easier to disseminate DE inferred on protein summaries. Moreover, MSqRobSum provides a highly modular analysis framework, which provides researchers with full flexibility to develop data analysis workflows tailored toward their specific applications.
基于无标记定量质谱的差异表达(DE)分析蛋白质的工作流程对数据分析提出了重要挑战,因为肽特异性效应和肽强度的上下文缺失。基于肽的工作流程,如 MSqRob,直接从肽强度中测试 DE,并优于首先将 MS1 肽强度汇总到蛋白质强度再进行 DE 分析的汇总方法。然而,这些方法计算成本高,对于非专业的终端用户来说往往难以理解,并且不提供蛋白质摘要,这对于可视化或下游处理很重要。因此,在这项工作中,我们使用基准 spike-in 数据集评估了最先进的汇总策略,并讨论了与基于肽的最先进模型 MSqRob 相比,这些策略失败的原因和时间。基于此评估,我们提出了一种新的汇总策略 MSqRobSum,它通过两步过程估计 MSqRob 的模型参数,避免了基于肽的工作流程的缺点。MSqRobSum 保持了 MSqRob 的卓越性能,同时为绘图和下游分析提供了有用的蛋白质表达摘要。将肽汇总到蛋白质强度可以大大降低计算复杂度、内存占用和模型复杂度,并使推断出的蛋白质摘要上的 DE 更容易传播。此外,MSqRobSum 提供了一个高度模块化的分析框架,为研究人员提供了充分的灵活性,以开发针对其特定应用的数据分析工作流程。