Wang Aiping, Xiao Yangyang, Huang Peng, Liu Lingjuan, Xiong Jie, Li Jian, Mao Ding'an, Liu Liqun
Department of Pediatrics, The Second Xiangya Hospital, Central South University, Changsha, China.
Department of Pediatrics Neurology, Children's Medical Center, The Second Xiangya Hospital, Central South University, Changsha, China.
Front Neurol. 2020 Apr 9;11:239. doi: 10.3389/fneur.2020.00239. eCollection 2020.
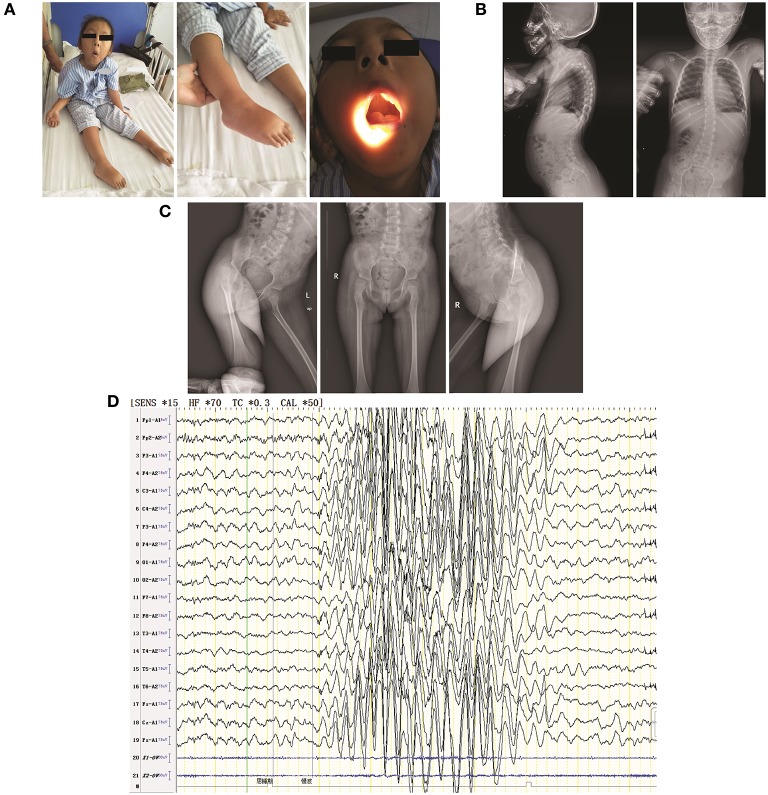
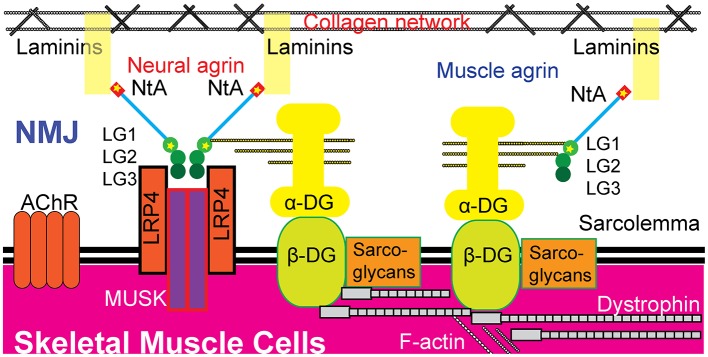
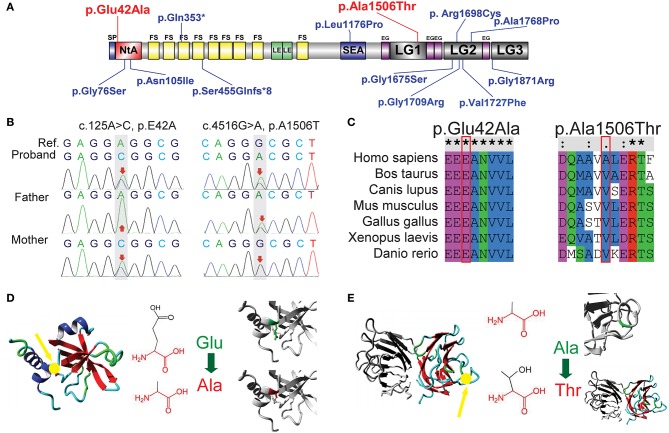
Congenital myasthenic syndrome (CMS) is a group of genetic disorders of neuromuscular transmission that is characterized by muscle weakness. A mutation in the gene encoding agrin () is a rare cause of CMS, and only a few families or isolated cases have been reported. We reported a pediatric proband exhibiting muscle weakness in the trunk and limbs with skeletal malformation and intellectual disability and performed whole-exome sequencing (WES) of the proband parent-offspring trio. Results revealed a new compound heterozygous mutation in : c.125A>C (p.Glu42Ala) in the N-terminal agrin domain (NtA) and c.4516G>A (p.Ala1506Thr) in the laminin G1 domain (LG1). Bioinformatic analysis predicted the mutation as possibly pathogenic. The new compound heterozygous mutation in may disrupt agrin's known function of bridging laminin and α-dystroglycan and undermine the formation and maintenance of the neuromuscular junction (NMJ) via both muscular and neural agrin pathways. It may also induce secondary peripheral neuropathy and skeletal malformation.
先天性肌无力综合征(CMS)是一组以肌肉无力为特征的神经肌肉传递遗传性疾病。编码集聚蛋白(agrin)的基因突变是CMS的罕见病因,仅报道过少数家族或散发病例。我们报告了一名表现为躯干和四肢肌肉无力并伴有骨骼畸形和智力残疾的儿科先证者,并对先证者及其父母进行了全外显子组测序(WES)。结果显示agrin基因存在一种新的复合杂合突变:N端集聚蛋白结构域(NtA)中的c.125A>C(p.Glu42Ala)以及层粘连蛋白G1结构域(LG1)中的c.4516G>A(p.Ala1506Thr)。生物信息学分析预测该突变可能具有致病性。agrin基因中的这种新的复合杂合突变可能会破坏集聚蛋白已知的连接层粘连蛋白和α- dystroglycan的功能,并通过肌肉和神经集聚蛋白途径破坏神经肌肉接头(NMJ)的形成和维持。它还可能诱发继发性周围神经病变和骨骼畸形。