Pathophysiology and Genetics of Neuron and Muscle, Faculté de Médecine Lyon Est, CNRS UMR 5261, INSERM U1315, Université Lyon1, Lyon, France.
Hospices Civils de Lyon, Groupement Est, Bron, France.
Acta Neuropathol. 2022 Oct;144(4):707-731. doi: 10.1007/s00401-022-02475-8. Epub 2022 Aug 10.
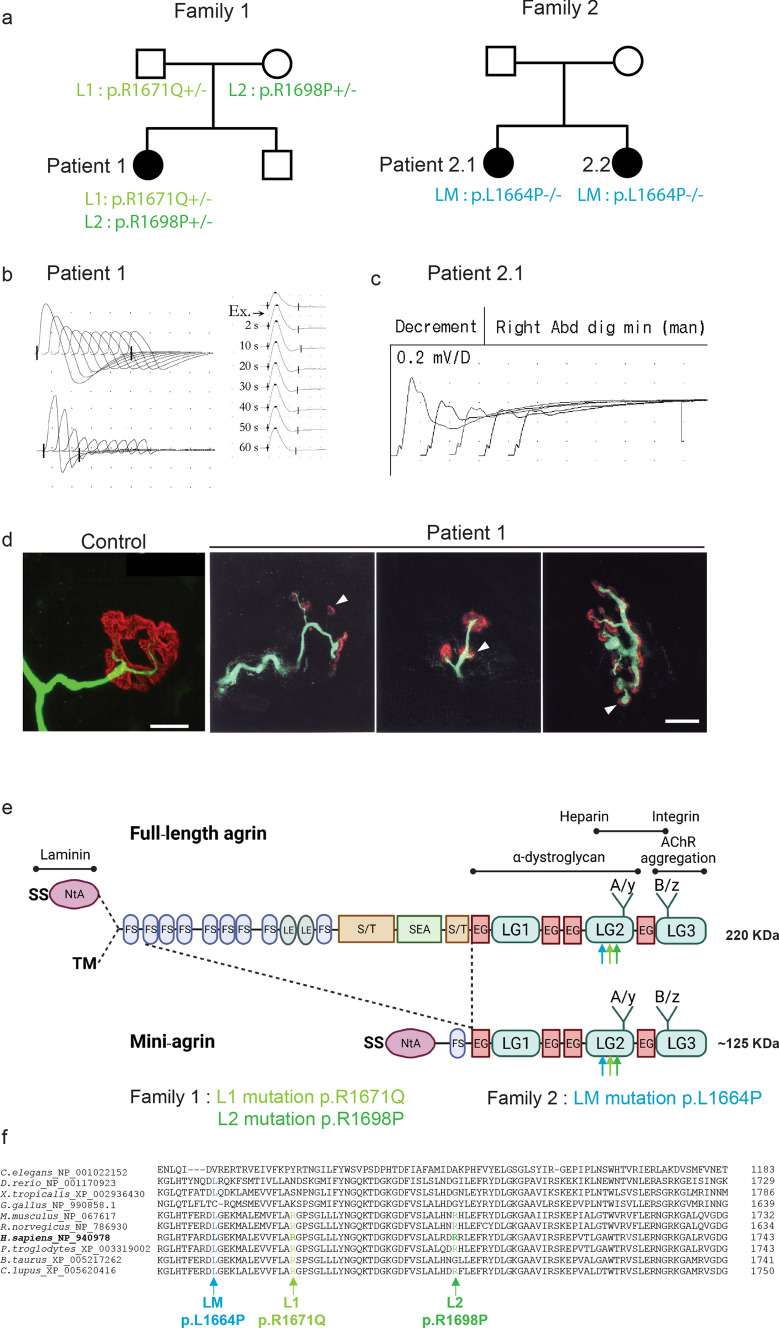
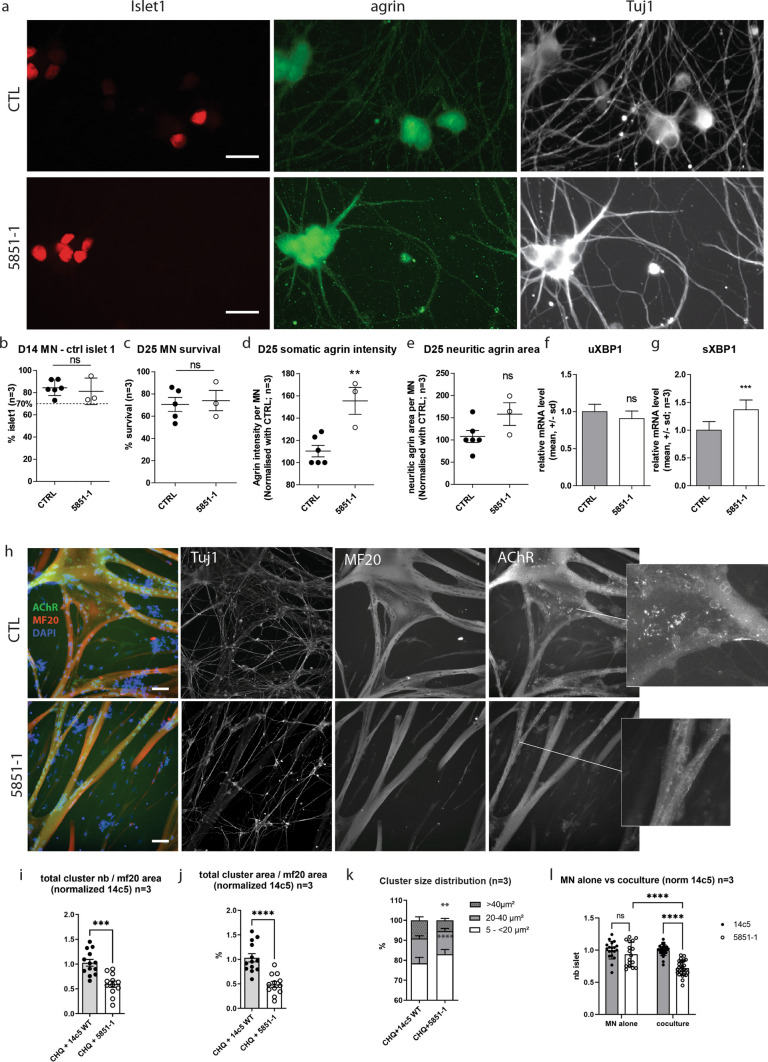
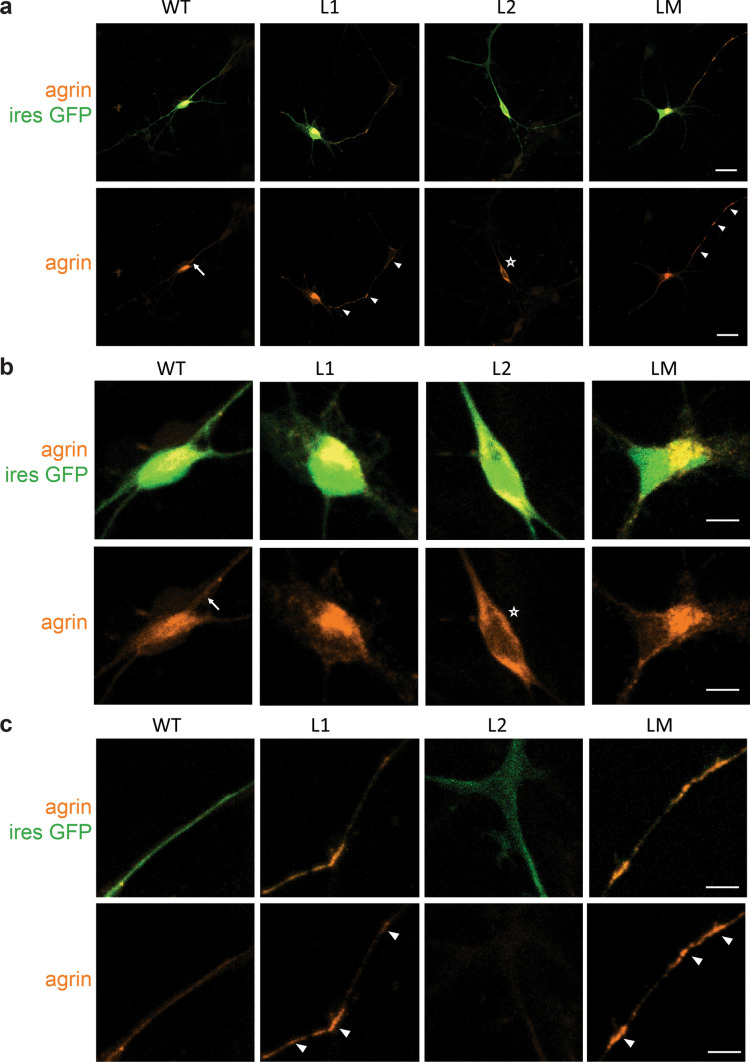
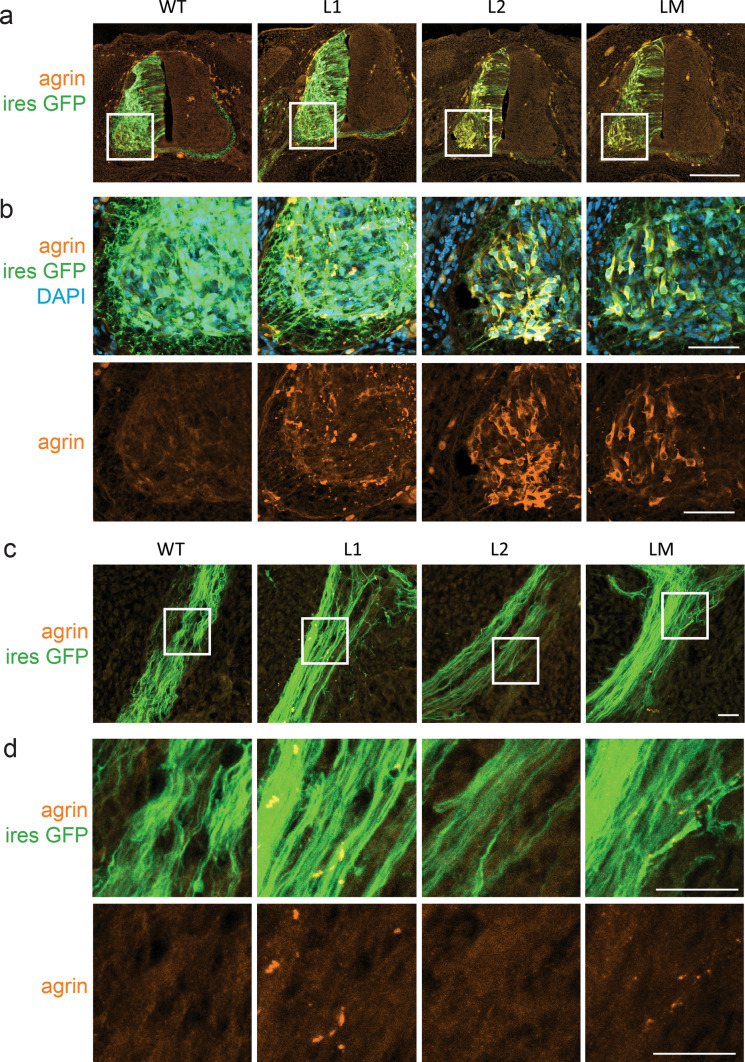
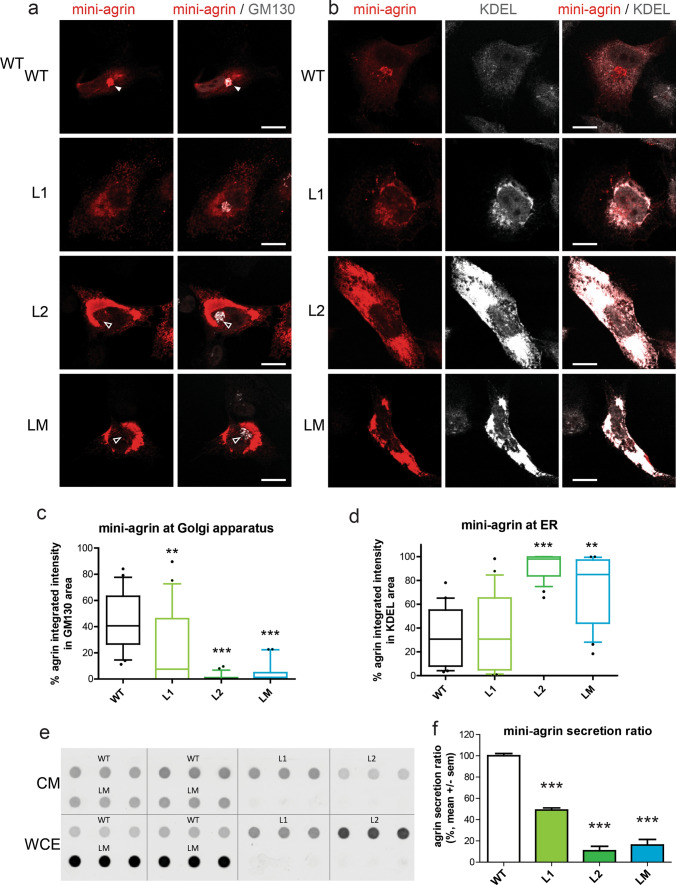
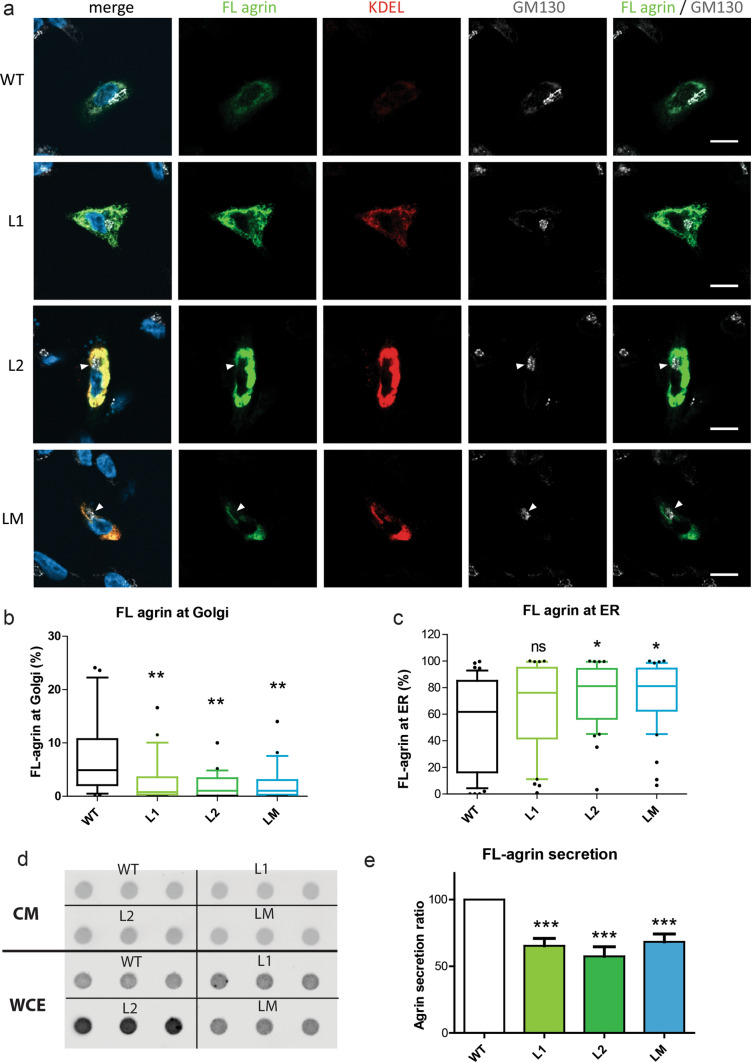
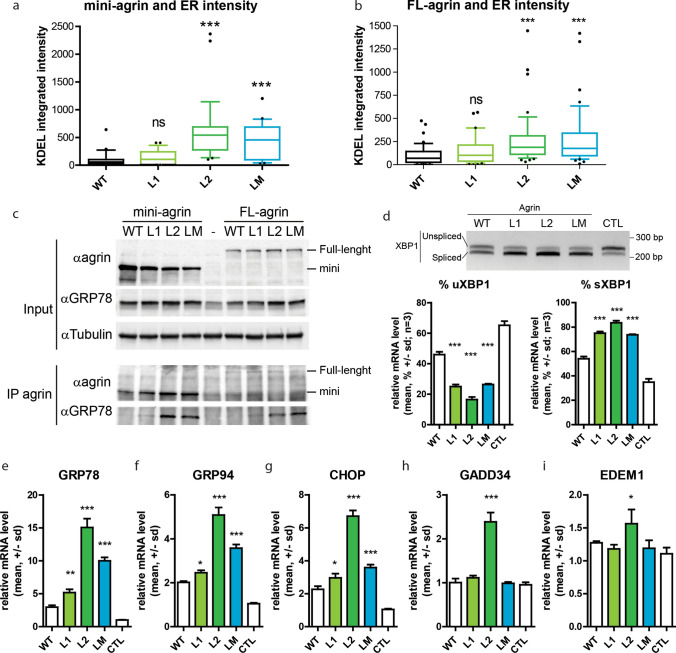
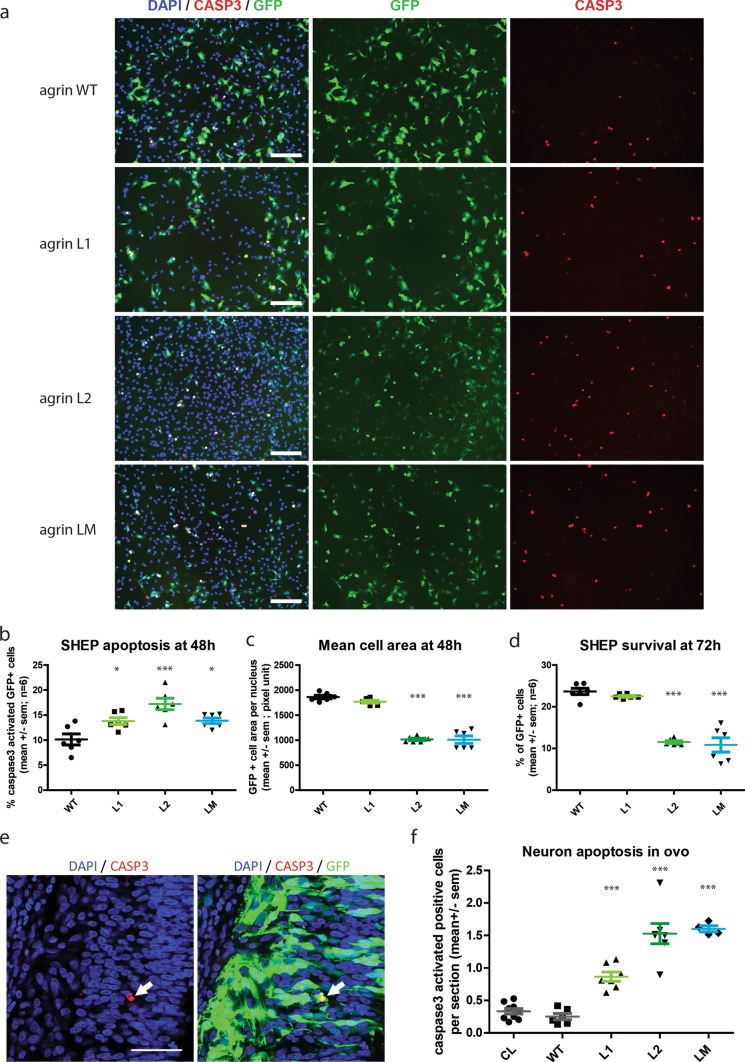
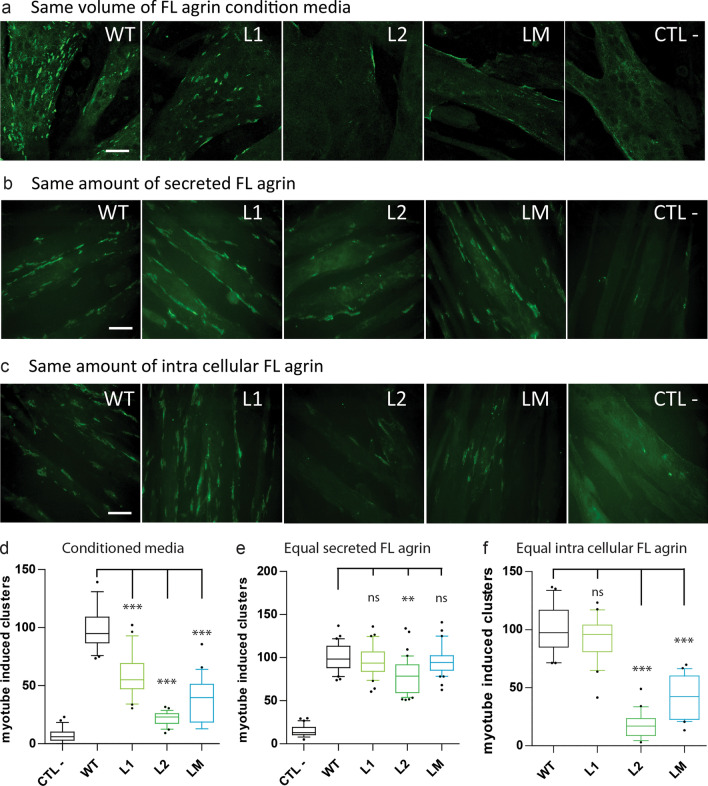
Congenital myasthenic syndromes (CMS) are predominantly characterized by muscle weakness and fatigability and can be caused by a variety of mutations in genes required for neuromuscular junction formation and maintenance. Among them, AGRN encodes agrin, an essential synaptic protein secreted by motoneurons. We have identified severe CMS patients with uncharacterized p.R1671Q, p.R1698P and p.L1664P mutations in the LG2 domain of agrin. Overexpression in primary motoneurons cultures in vitro and in chick spinal motoneurons in vivo revealed that the mutations modified agrin trafficking, leading to its accumulation in the soma and/or in the axon. Expression of mutant agrins in cultured cells demonstrated accumulation of agrin in the endoplasmic reticulum associated with induction of unfolded protein response (UPR) and impaired secretion in the culture medium. Interestingly, evaluation of the specific activity of individual agrins on AChR cluster formation indicated that when secreted, mutant agrins retained a normal capacity to trigger the formation of AChR clusters. To confirm agrin accumulation and secretion defect, iPS cells were derived from a patient and differentiated into motoneurons. Patient iPS-derived motoneurons accumulated mutant agrin in the soma and increased XBP1 mRNA splicing, suggesting UPR activation. Moreover, co-cultures of patient iPS-derived motoneurons with myotubes confirmed the deficit in agrin secretion and revealed a reduction in motoneuron survival. Altogether, we report the first mutations in AGRN gene that specifically affect agrin secretion by motoneurons. Interestingly, the three patients carrying these mutations were initially suspected of spinal muscular atrophy (SMA). Therefore, in the presence of patients with a clinical presentation of SMA but without mutation in the SMN1 gene, it can be worth to look for mutations in AGRN.
先天性肌无力综合征(CMS)主要表现为肌肉无力和易疲劳,可由神经肌肉接头形成和维持所需基因的多种突变引起。其中,AGRN 编码 agrin,一种运动神经元分泌的必需突触蛋白。我们在 agrin 的 LG2 结构域中发现了具有未表征的 p.R1671Q、p.R1698P 和 p.L1664P 突变的严重 CMS 患者。体外原代运动神经元培养和体内鸡脊髓运动神经元中的过表达表明,这些突变改变了 agrin 的运输,导致其在体和/或轴突中积累。在培养细胞中表达突变的 agrins 表明 agrin 在与未折叠蛋白反应(UPR)诱导和培养基中分泌受损相关的内质网中积累。有趣的是,评估突变的 agrins 在单个 AChR 簇形成上的特定活性表明,当分泌时,突变的 agrins 仍然具有触发 AChR 簇形成的正常能力。为了确认 agrin 积累和分泌缺陷,我们从患者中获得 iPS 细胞并分化为运动神经元。患者 iPS 衍生的运动神经元在体中积累突变的 agrin,并增加 XBP1 mRNA 剪接,表明 UPR 激活。此外,患者 iPS 衍生的运动神经元与肌管的共培养证实了 agrin 分泌缺陷,并揭示了运动神经元存活减少。总之,我们报告了第一个特定影响运动神经元 agrin 分泌的 AGRN 基因突变。有趣的是,携带这些突变的三名患者最初被怀疑患有脊髓性肌萎缩症(SMA)。因此,在存在具有 SMA 临床表现但 SMN1 基因无突变的患者中,值得寻找 AGRN 基因突变。