Hahn Katharina, Pollmann Liart, Nowak Juliette, Nguyen Ariane Hai Ha, Haake Kathrin, Neehus Anna-Lena, Waqas Syed F Hassnain, Pessler Frank, Baumann Ulrich, Hetzel Miriam, Casanova Jean-Laurent, Schulz Ansgar, Bustamante Jacinta, Ackermann Mania, Lachmann Nico
Translational Hematology of Congenital Diseases, Institute of Experimental Hematology, Hannover Medical School, Hannover, Germany.
REBIRTH Research Center for Translational and Regenerative Medicine, Hannover, Germany.
Mol Ther Methods Clin Dev. 2020 Apr 11;17:785-795. doi: 10.1016/j.omtm.2020.04.002. eCollection 2020 Jun 12.
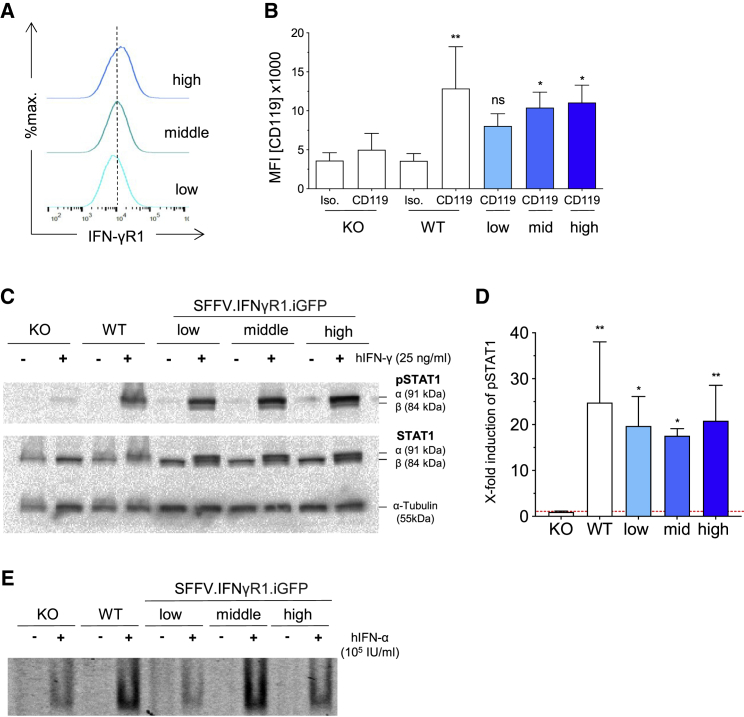
Autosomal recessive (AR) complete interferon-γ receptor 1 (IFN-γR1) deficiency, also known as one genetic etiology of Mendelian susceptibility to mycobacterial disease (MSMD), is a life-threatening congenital disease leading to premature death. Affected patients present a pathognomonic predisposition to recurrent and severe infections with environmental mycobacteria or the bacillus Calmette-Guérin (BCG) vaccine. Current therapeutic options are limited to antibiotic treatment and hematopoietic stem cell transplantation, however with poor outcome. Given the clinical success of gene therapy, we introduce the first lentiviral-based gene therapy approach to restore expression and function of the human IFN-γR-downstream signaling cascade. In our study, we developed lentiviral vectors constitutively expressing the human IFN-γR1 and demonstrate stable transgene expression without interference with cell viability and proliferation in transduced human hematopoietic cells. Using an IFN-γR1-deficient HeLa cell model, we show stable receptor reconstitution and restored IFN-γR1 signaling without adverse effect on cell functionality. Transduction of both SV40-immortalized and primary fibroblasts derived from IFN-γR1-deficient MSMD patients was able to recover IFN-γR1 expression and restore type II IFN signaling upon stimulation with IFN-γ. In summary, we highlight lentiviral vectors to correct the IFN-γ mediated immunity and present the first gene therapy approach for patients suffering from AR complete IFN-γR1 deficiency.
常染色体隐性(AR)完全性γ干扰素受体1(IFN-γR1)缺陷,也被称为孟德尔易感性分枝杆菌病(MSMD)的一种遗传病因,是一种危及生命的先天性疾病,可导致过早死亡。受影响的患者对环境分枝杆菌或卡介苗(BCG)疫苗反复发生严重感染呈现出特征性的易感性。目前的治疗选择仅限于抗生素治疗和造血干细胞移植,然而效果不佳。鉴于基因治疗在临床上的成功,我们引入了第一种基于慢病毒的基因治疗方法,以恢复人类IFN-γ下游信号级联反应的表达和功能。在我们的研究中,我们开发了组成性表达人类IFN-γR1的慢病毒载体,并证明在转导的人类造血细胞中,转基因表达稳定,且不干扰细胞活力和增殖。使用IFN-γR1缺陷的HeLa细胞模型,我们展示了稳定的受体重构以及恢复的IFN-γR1信号传导,且对细胞功能没有不利影响。对来自IFN-γR1缺陷的MSMD患者的SV40永生化和成纤维原代细胞进行转导,能够在受到IFN-γ刺激后恢复IFN-γR1表达并恢复II型干扰素信号传导。总之,我们强调慢病毒载体可纠正IFN-γ介导的免疫,并为患有AR完全性IFN-γR1缺陷的患者提出了第一种基因治疗方法。