UNC Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC, USA.
Chem Soc Rev. 2020 Jun 7;49(11):3525-3564. doi: 10.1039/d0cs00098a. Epub 2020 May 1.
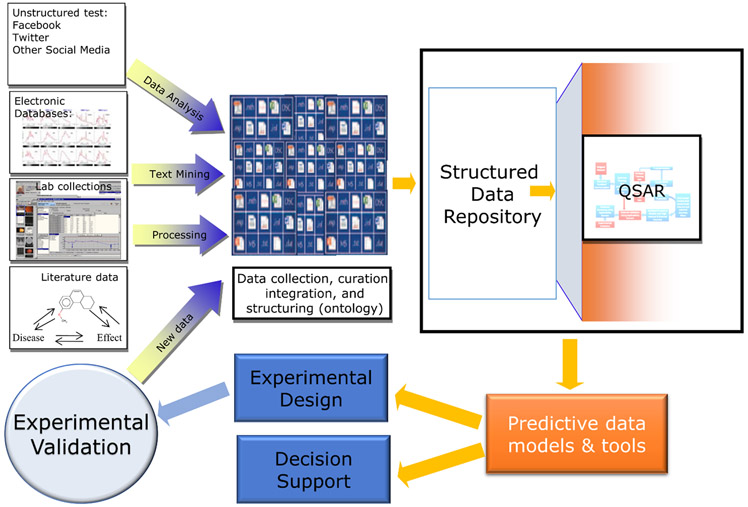
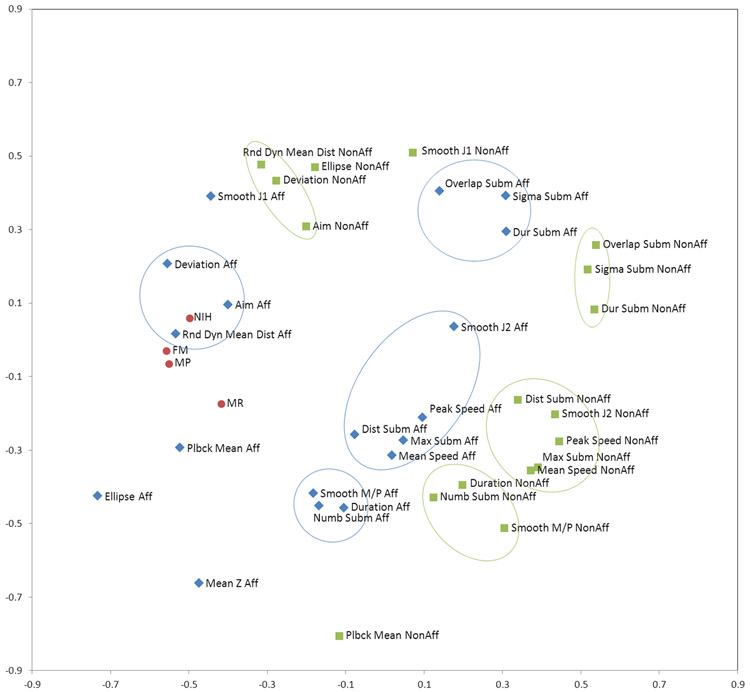
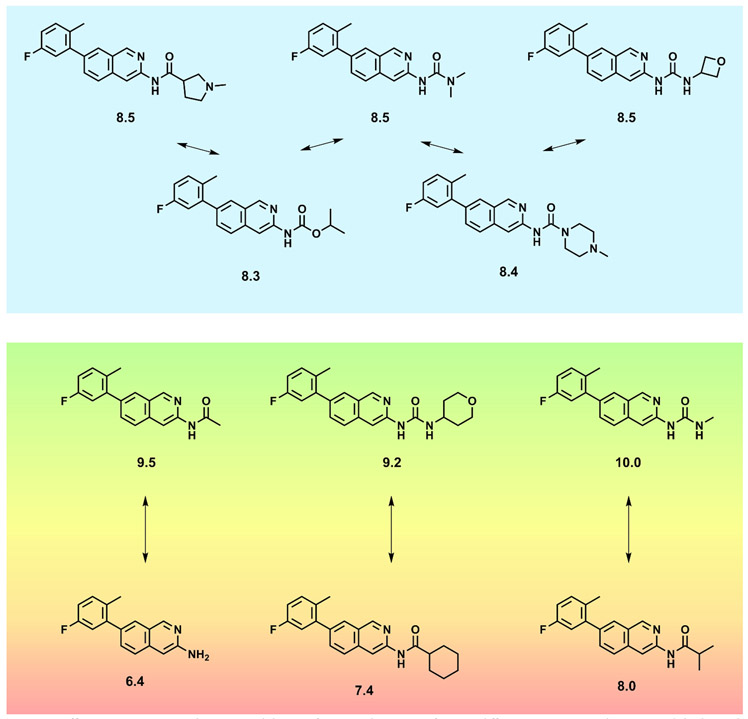
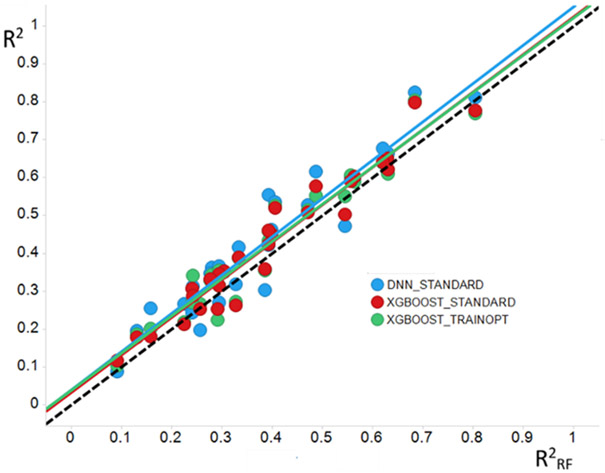
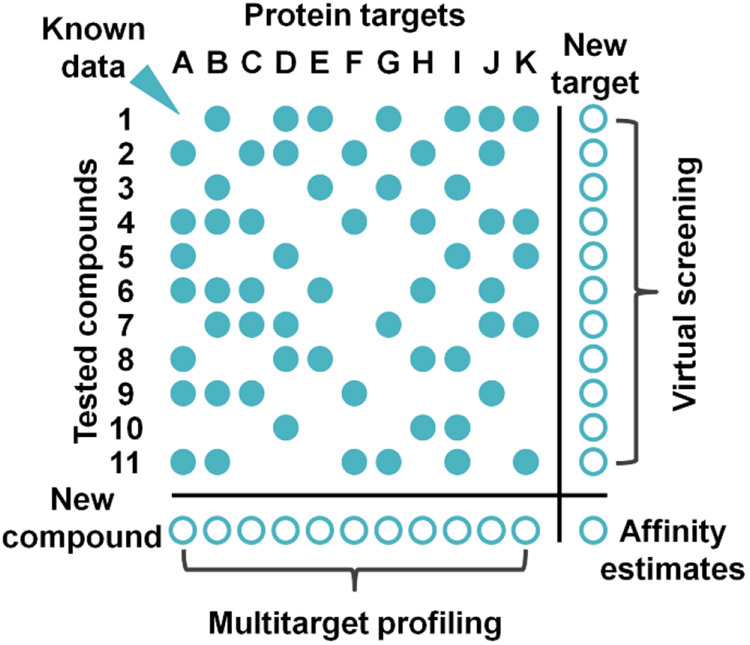
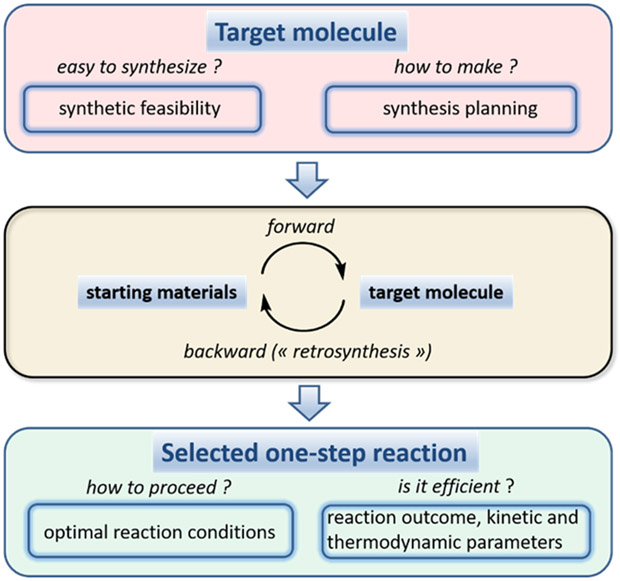
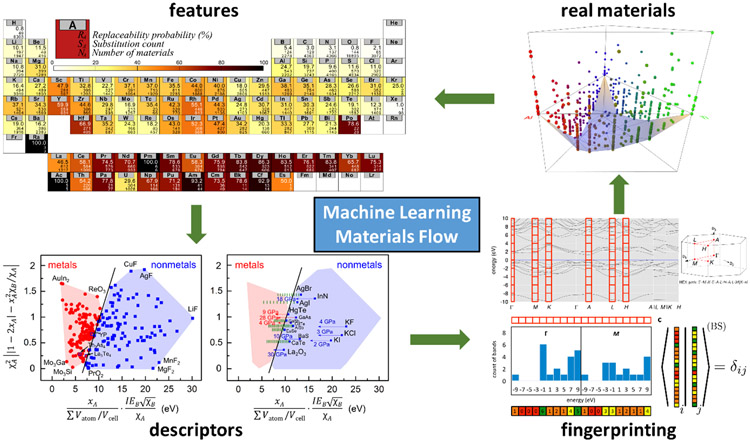
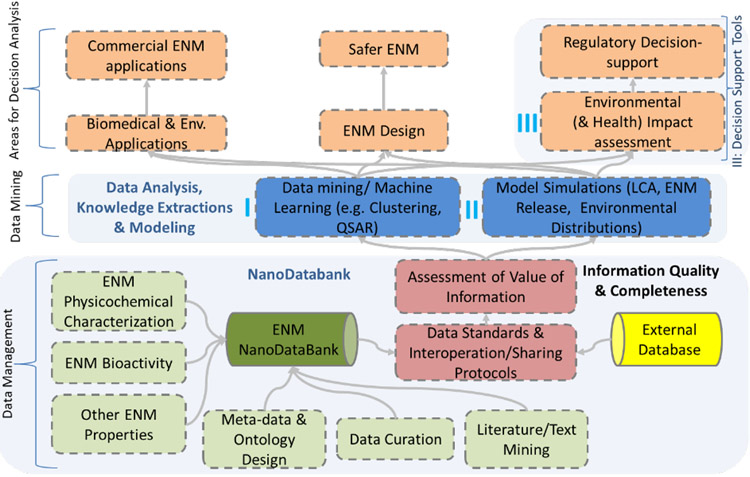
Prediction of chemical bioactivity and physical properties has been one of the most important applications of statistical and more recently, machine learning and artificial intelligence methods in chemical sciences. This field of research, broadly known as quantitative structure-activity relationships (QSAR) modeling, has developed many important algorithms and has found a broad range of applications in physical organic and medicinal chemistry in the past 55+ years. This Perspective summarizes recent technological advances in QSAR modeling but it also highlights the applicability of algorithms, modeling methods, and validation practices developed in QSAR to a wide range of research areas outside of traditional QSAR boundaries including synthesis planning, nanotechnology, materials science, biomaterials, and clinical informatics. As modern research methods generate rapidly increasing amounts of data, the knowledge of robust data-driven modelling methods professed within the QSAR field can become essential for scientists working both within and outside of chemical research. We hope that this contribution highlighting the generalizable components of QSAR modeling will serve to address this challenge.
化学生物活性和物理性质的预测一直是统计方法,最近更是机器学习和人工智能方法在化学科学中最重要的应用之一。这个研究领域,通常被称为定量构效关系(QSAR)建模,在过去的 55 年以上的时间里,已经开发出了许多重要的算法,并在物理有机化学和药物化学中找到了广泛的应用。本文综述了 QSAR 建模的最新技术进展,但也强调了在传统 QSAR 边界之外的广泛研究领域(包括合成规划、纳米技术、材料科学、生物材料和临床信息学)中开发的算法、建模方法和验证实践的适用性。随着现代研究方法生成的数据量迅速增加,QSAR 领域中公认的稳健数据驱动建模方法的知识对于在化学研究内外工作的科学家来说可能变得至关重要。我们希望,强调 QSAR 建模的可推广部分的这一贡献将有助于应对这一挑战。