CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Lazarettgasse 14, AKH BT 25.3, 1090, Vienna, Austria.
Department of Dermatology, Medical University of Vienna, 1090, Vienna, Austria.
Genome Biol. 2020 May 6;21(1):106. doi: 10.1186/s13059-020-02006-2.
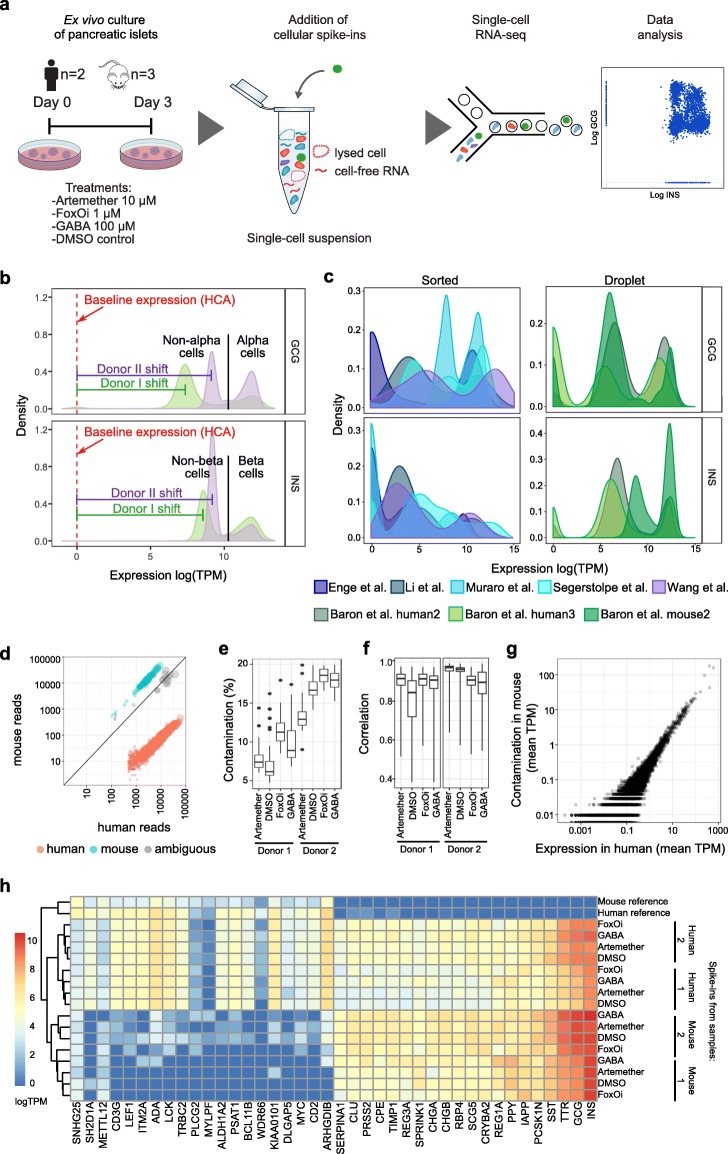
Single-cell RNA-seq (scRNA-seq) is emerging as a powerful tool to dissect cell-specific effects of drug treatment in complex tissues. This application requires high levels of precision, robustness, and quantitative accuracy-beyond those achievable with existing methods for mainly qualitative single-cell analysis. Here, we establish the use of standardized reference cells as spike-in controls for accurate and robust dissection of single-cell drug responses.
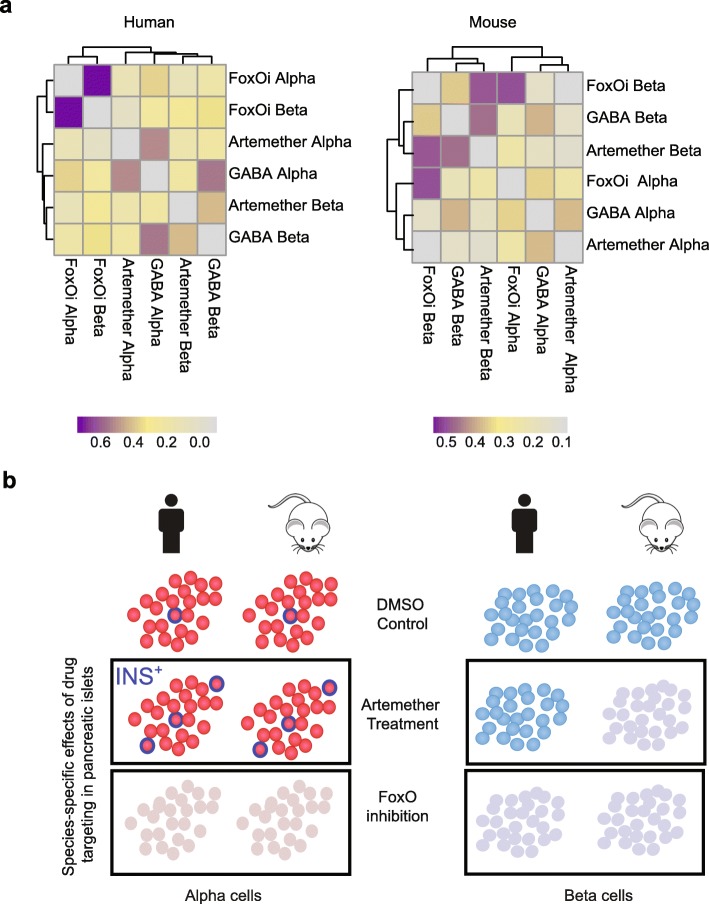
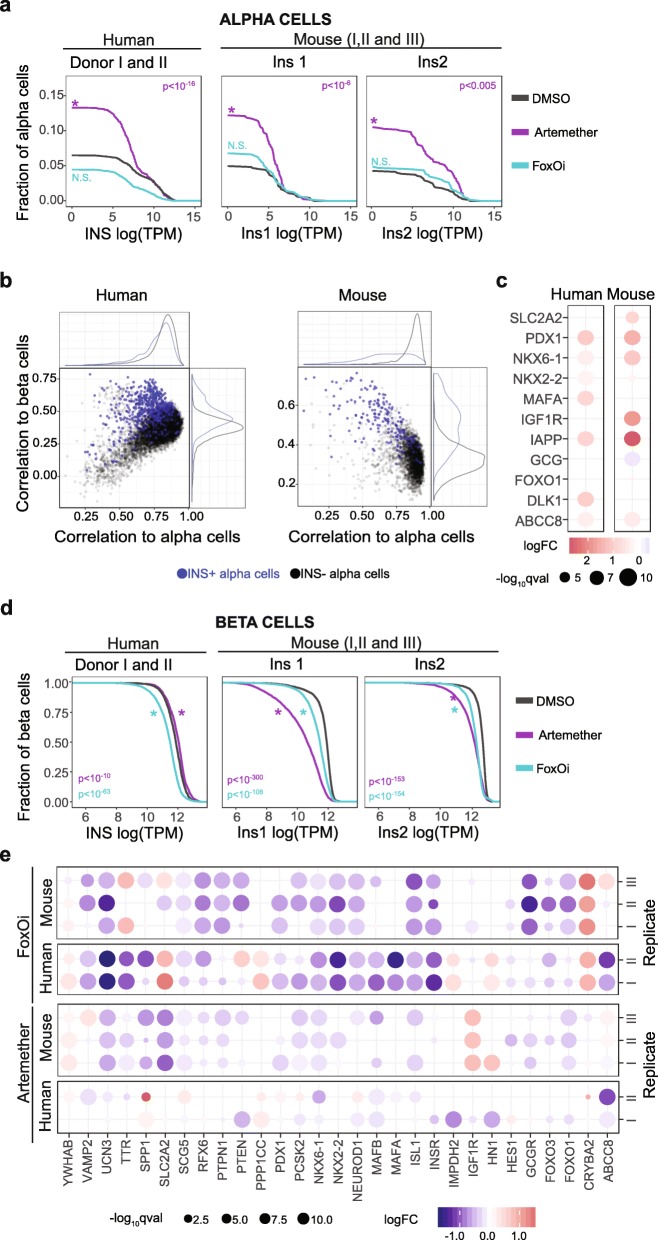
We find that contamination by cell-free RNA can constitute up to 20% of reads in human primary tissue samples, and we show that the ensuing biases can be removed effectively using a novel bioinformatics algorithm. Applying our method to both human and mouse pancreatic islets treated ex vivo, we obtain an accurate and quantitative assessment of cell-specific drug effects on the transcriptome. We observe that FOXO inhibition induces dedifferentiation of both alpha and beta cells, while artemether treatment upregulates insulin and other beta cell marker genes in a subset of alpha cells. In beta cells, dedifferentiation and insulin repression upon artemether treatment occurs predominantly in mouse but not in human samples.
This new method for quantitative, error-correcting, scRNA-seq data normalization using spike-in reference cells helps clarify complex cell-specific effects of pharmacological perturbations with single-cell resolution and high quantitative accuracy.
单细胞 RNA 测序(scRNA-seq)正成为剖析复杂组织中药物处理对细胞特异性影响的强大工具。这种应用需要高精度、高稳健性和定量准确性——超越现有主要用于单细胞定性分析的方法所能达到的水平。在这里,我们建立了使用标准化参考细胞作为内参的方法,以准确稳健地剖析单细胞药物反应。
我们发现,无细胞 RNA 的污染可达人类原代组织样本中读取序列的 20%,我们表明,通过一种新的生物信息学算法可以有效地去除由此产生的偏差。将我们的方法应用于体外处理的人和小鼠胰岛,我们对转录组上药物对细胞特异性的影响进行了准确和定量的评估。我们观察到 FOXO 抑制诱导 alpha 和 beta 细胞的去分化,而青蒿素处理在上调 alpha 细胞中胰岛素和其他 beta 细胞标记基因的同时诱导 beta 细胞的去分化和胰岛素抑制。在 beta 细胞中,青蒿素处理后的去分化和胰岛素抑制主要发生在小鼠而非人类样本中。
这种使用内参进行定量、纠错、scRNA-seq 数据标准化的新方法有助于以单细胞分辨率和高定量准确性阐明药理学扰动的复杂细胞特异性影响。