Department of Radiation Oncology, Perlmutter Cancer Center, New York University School of Medicine, New York, NY, USA.
Department of Anatomy, University of California, San Francisco, San Francisco, CA, USA.
Nature. 2020 May;581(7806):100-105. doi: 10.1038/s41586-020-2229-5. Epub 2020 Apr 22.
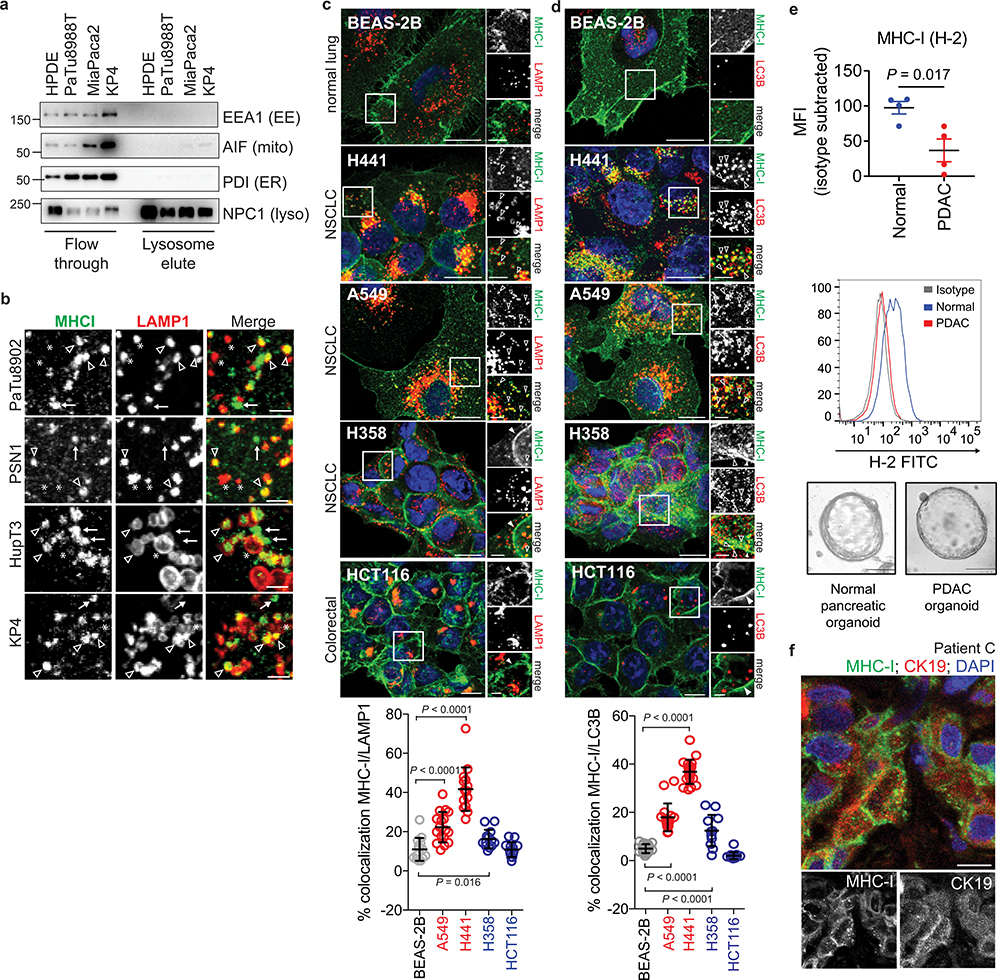
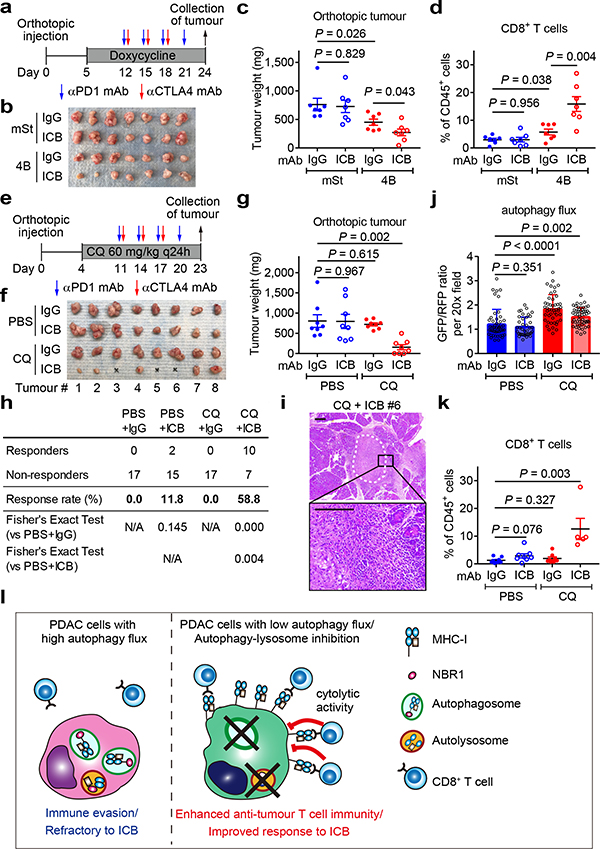
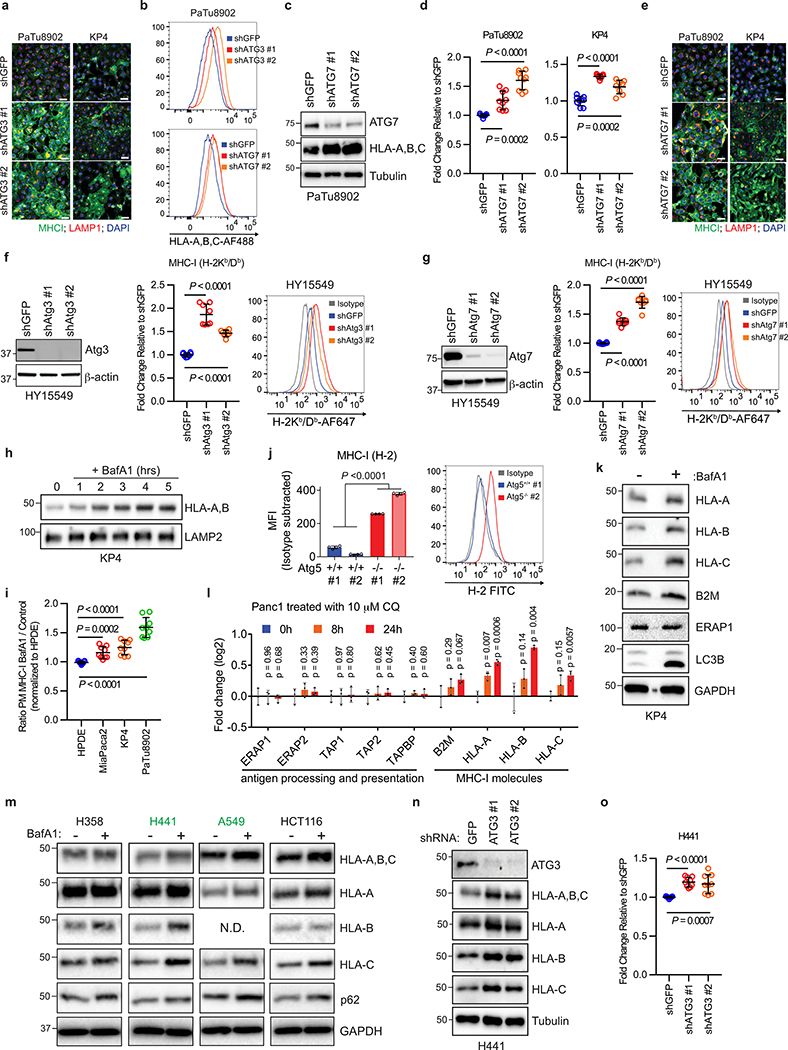
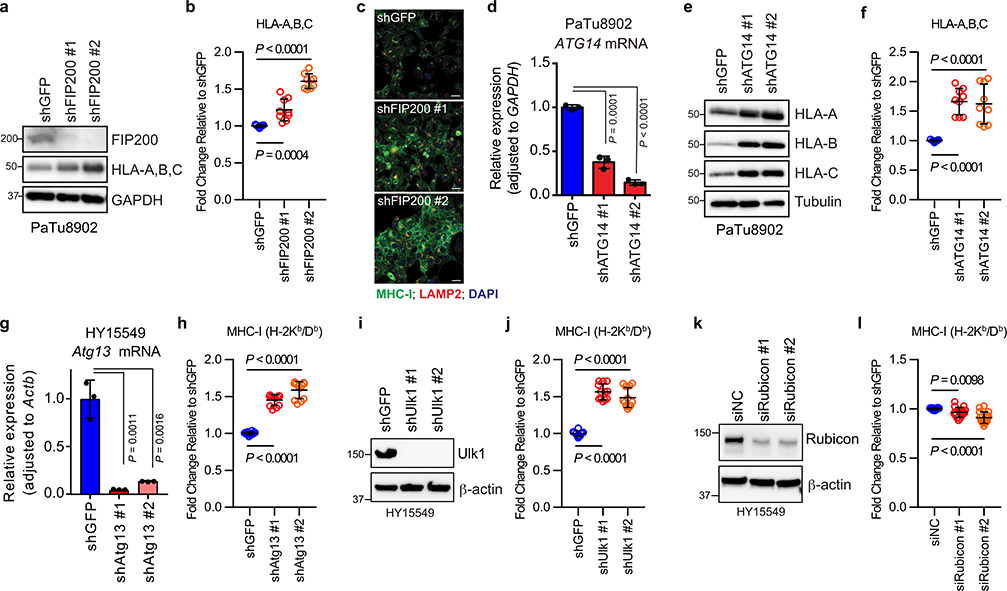
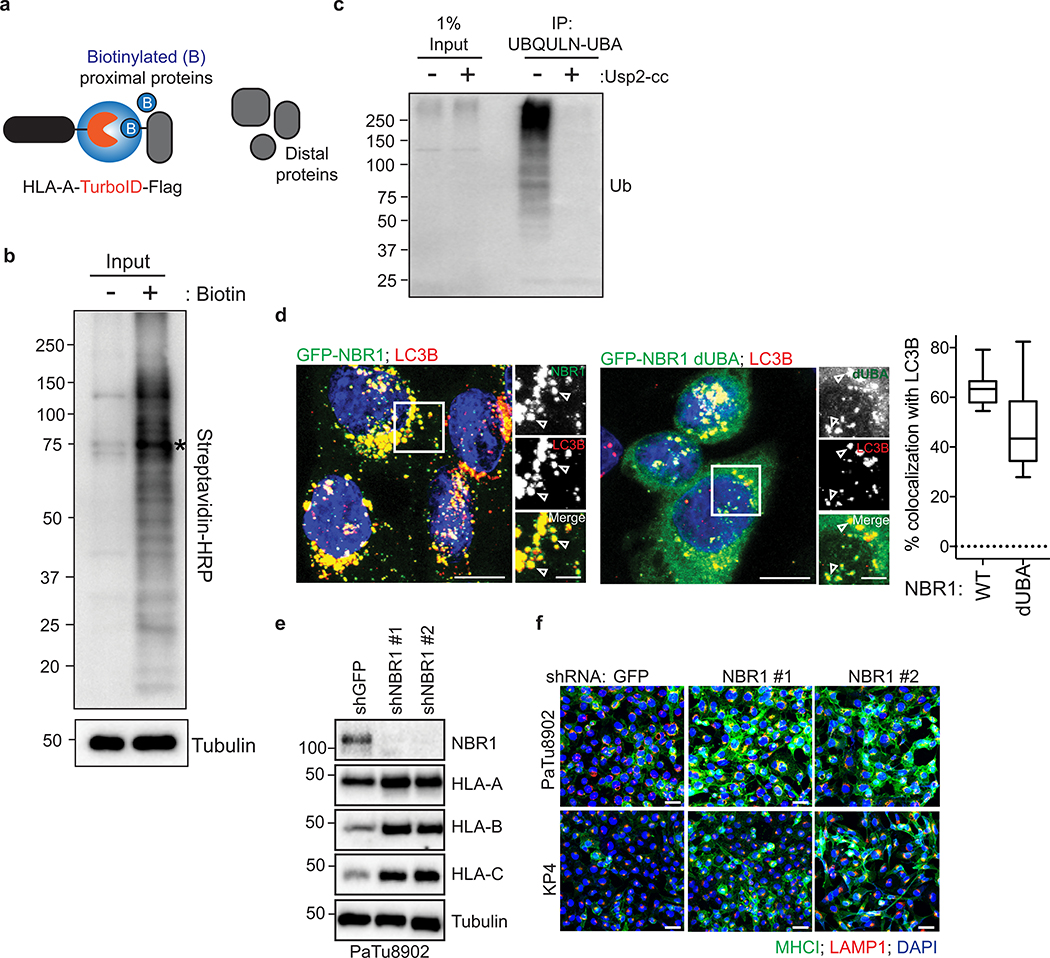
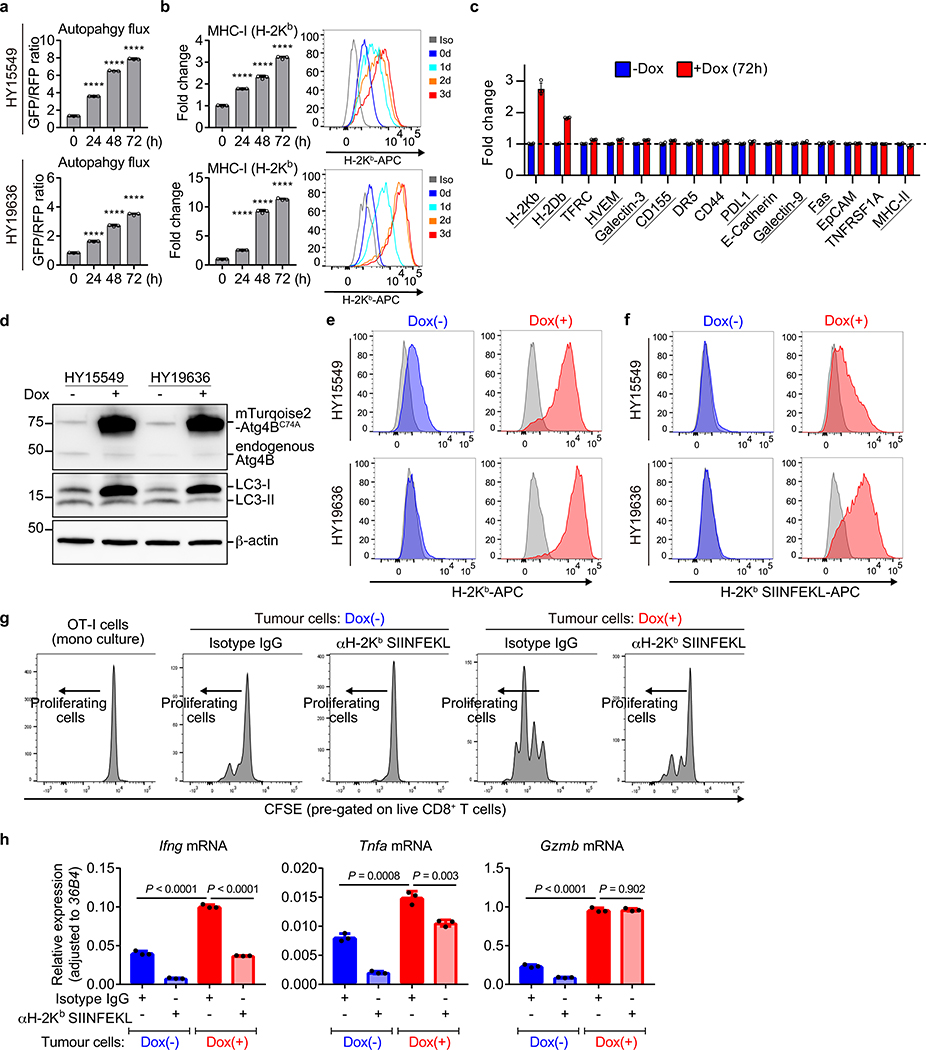
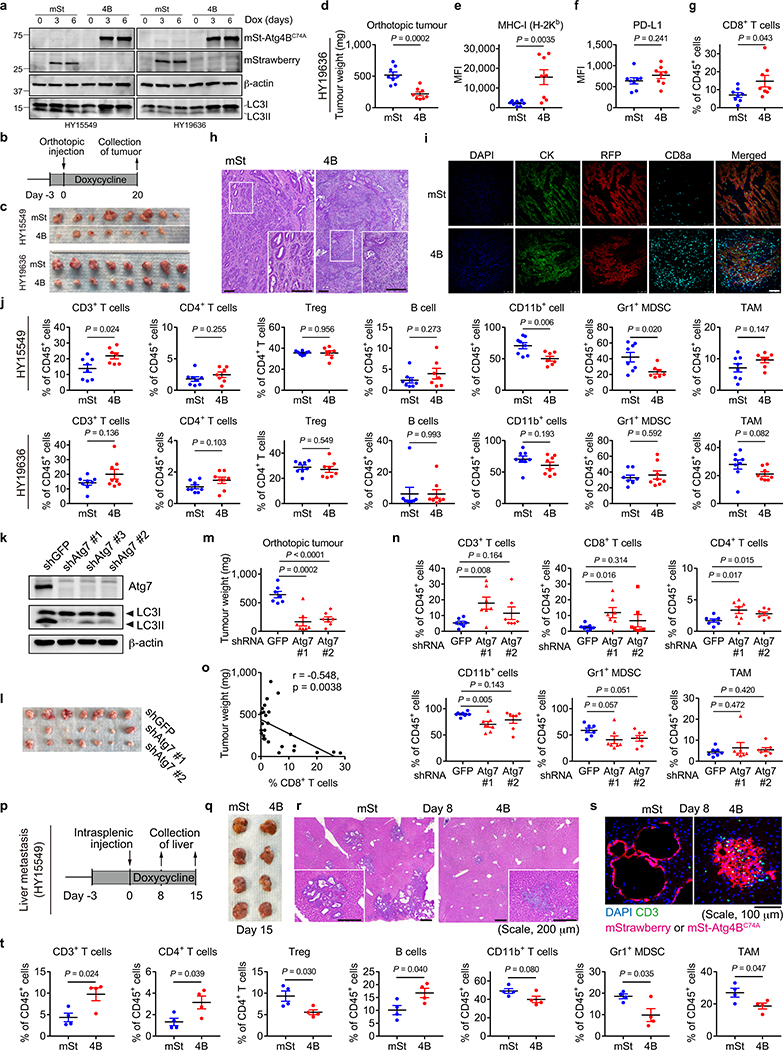
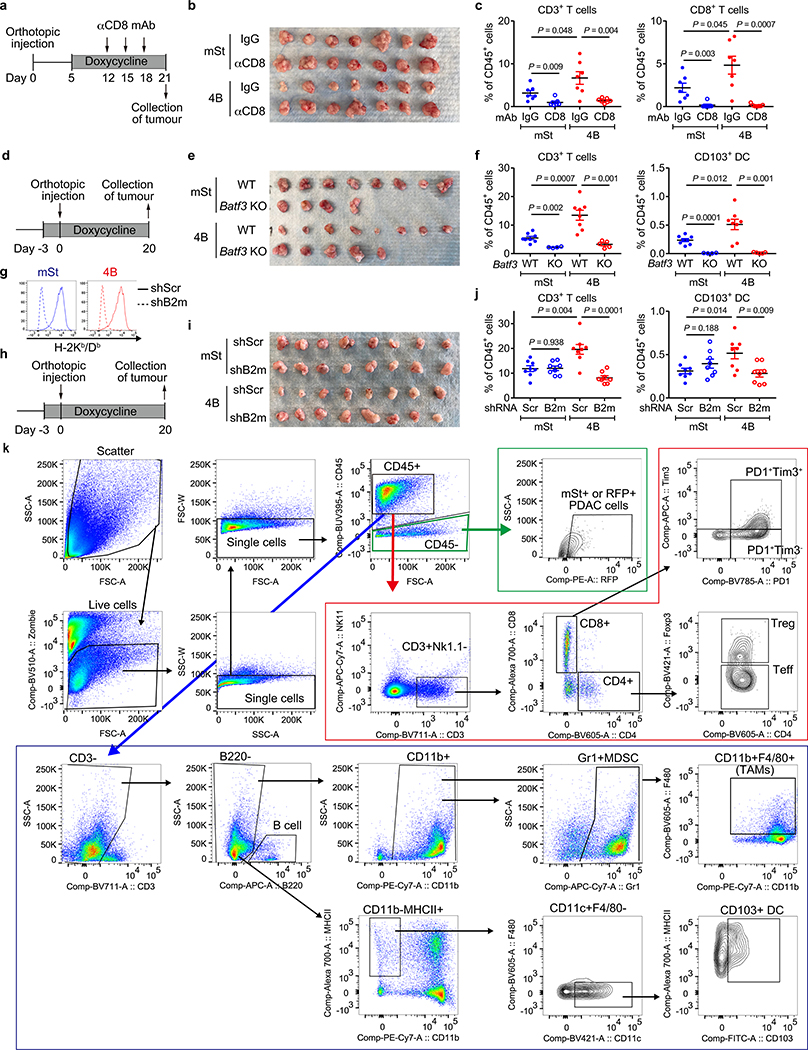
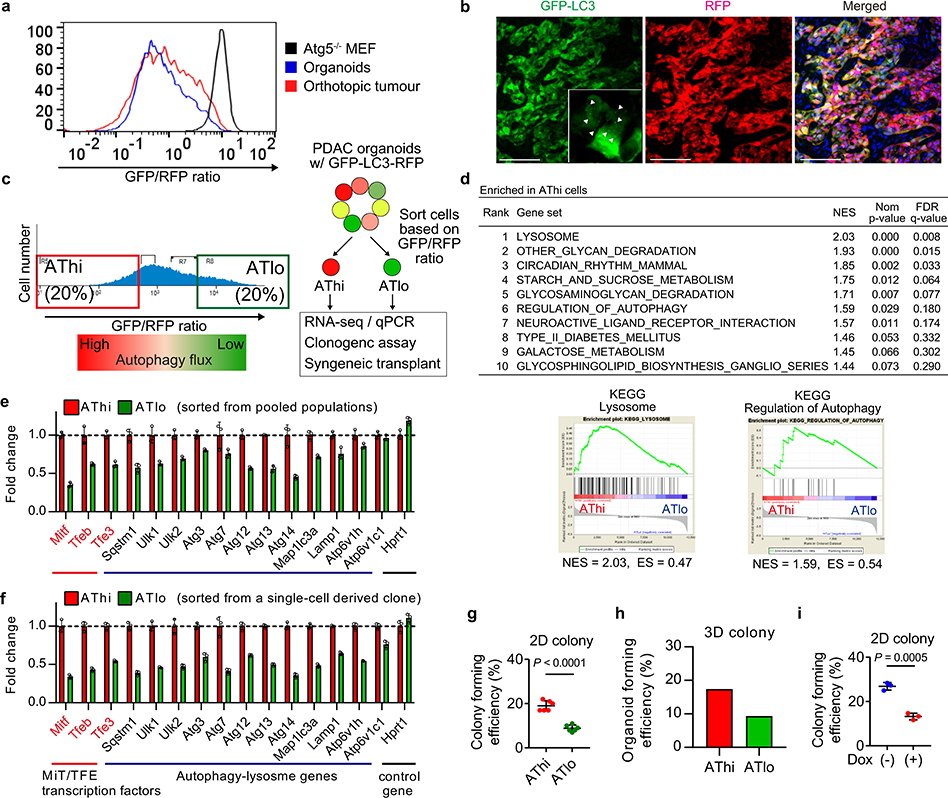
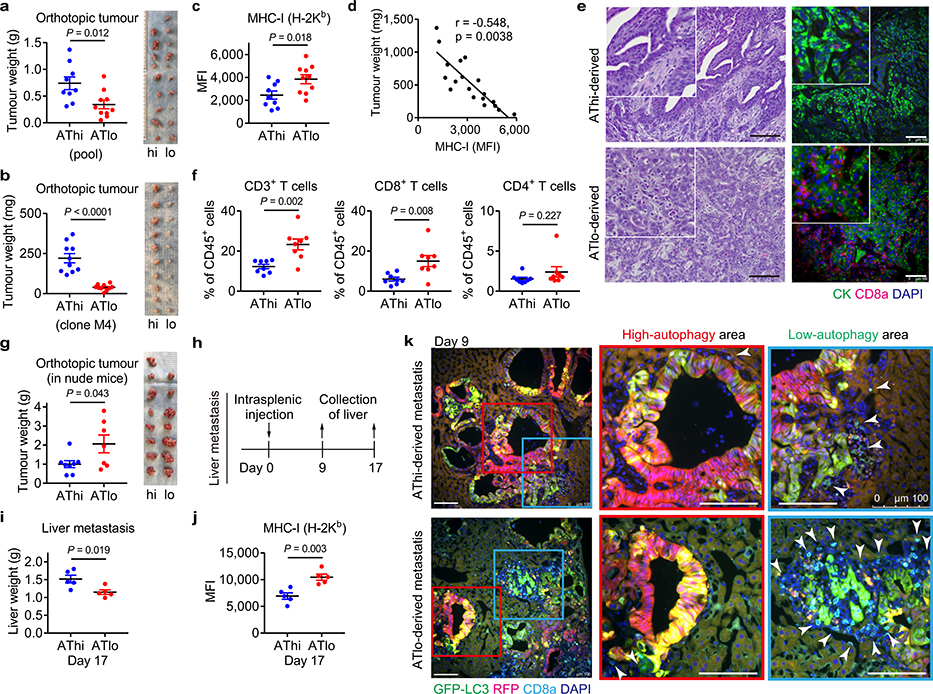
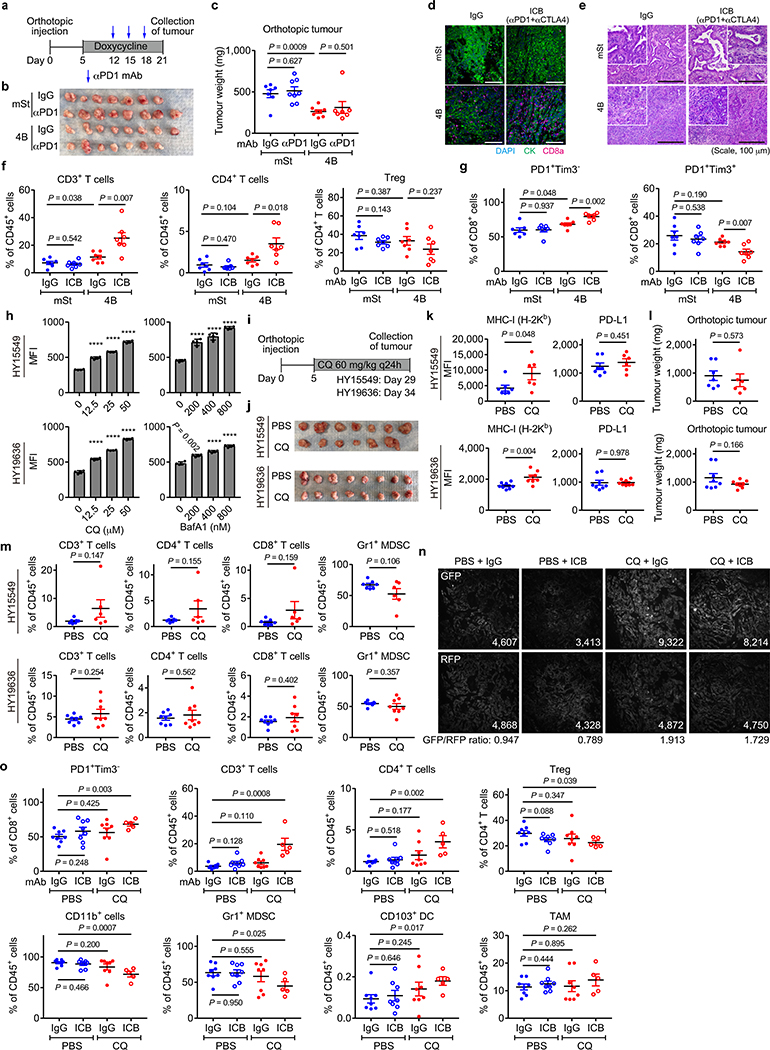
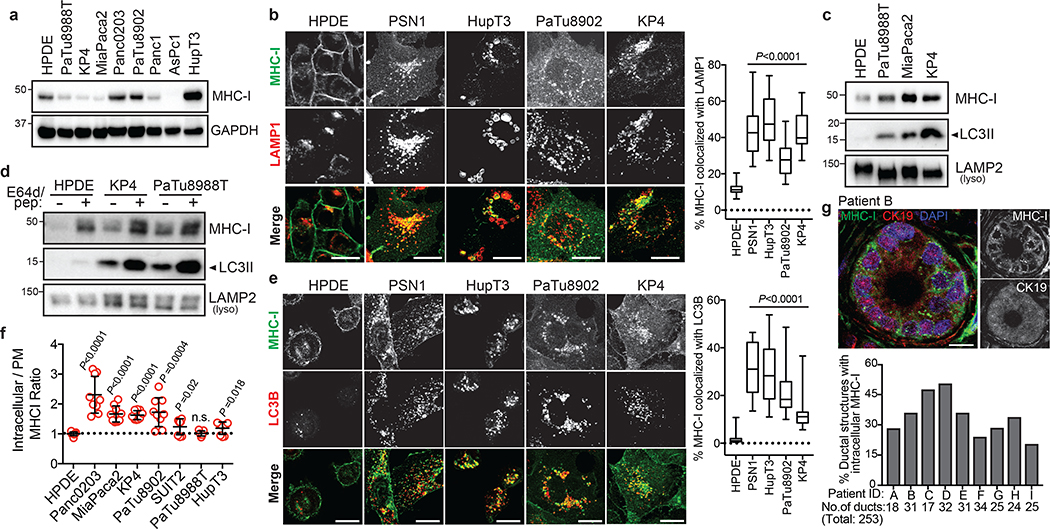
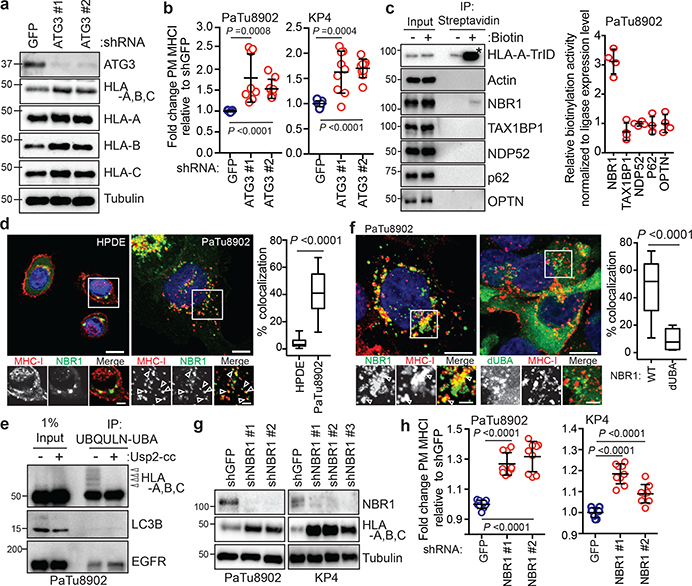
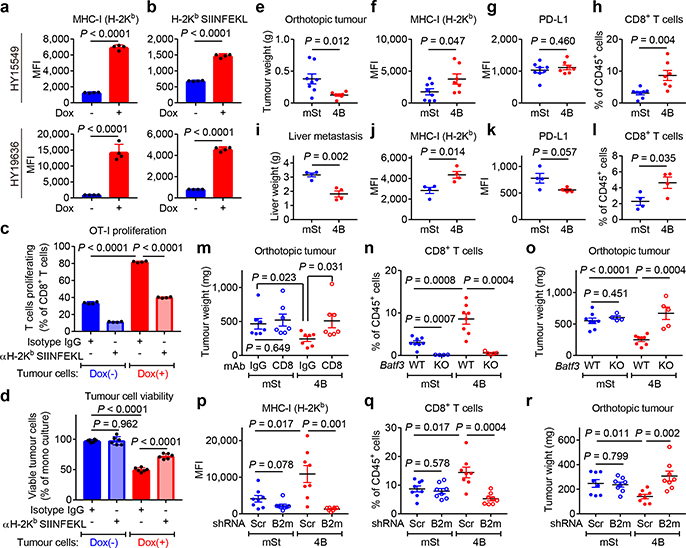
Immune evasion is a major obstacle for cancer treatment. Common mechanisms of evasion include impaired antigen presentation caused by mutations or loss of heterozygosity of the major histocompatibility complex class I (MHC-I), which has been implicated in resistance to immune checkpoint blockade (ICB) therapy. However, in pancreatic ductal adenocarcinoma (PDAC), which is resistant to most therapies including ICB, mutations that cause loss of MHC-I are rarely found despite the frequent downregulation of MHC-I expression. Here we show that, in PDAC, MHC-I molecules are selectively targeted for lysosomal degradation by an autophagy-dependent mechanism that involves the autophagy cargo receptor NBR1. PDAC cells display reduced expression of MHC-I at the cell surface and instead demonstrate predominant localization within autophagosomes and lysosomes. Notably, inhibition of autophagy restores surface levels of MHC-I and leads to improved antigen presentation, enhanced anti-tumour T cell responses and reduced tumour growth in syngeneic host mice. Accordingly, the anti-tumour effects of autophagy inhibition are reversed by depleting CD8 T cells or reducing surface expression of MHC-I. Inhibition of autophagy, either genetically or pharmacologically with chloroquine, synergizes with dual ICB therapy (anti-PD1 and anti-CTLA4 antibodies), and leads to an enhanced anti-tumour immune response. Our findings demonstrate a role for enhanced autophagy or lysosome function in immune evasion by selective targeting of MHC-I molecules for degradation, and provide a rationale for the combination of autophagy inhibition and dual ICB therapy as a therapeutic strategy against PDAC.
免疫逃逸是癌症治疗的主要障碍。常见的逃逸机制包括主要组织相容性复合体 I 类 (MHC-I) 的突变或杂合性丢失导致的抗原呈递受损,这与免疫检查点阻断 (ICB) 治疗的耐药性有关。然而,在对大多数治疗方法包括 ICB 都有耐药性的胰腺导管腺癌 (PDAC) 中,尽管 MHC-I 表达经常下调,但很少发现导致 MHC-I 丢失的突变。在这里,我们表明,在 PDAC 中,MHC-I 分子通过一种自噬依赖性机制被选择性靶向溶酶体降解,该机制涉及自噬货物受体 NBR1。PDAC 细胞表面 MHC-I 的表达减少,而是主要定位于自噬体和溶酶体中。值得注意的是,抑制自噬可恢复 MHC-I 的表面水平,并导致抗原呈递增强、抗肿瘤 T 细胞反应增强和同种异体宿主小鼠肿瘤生长减少。因此,自噬抑制的抗肿瘤作用可通过耗尽 CD8 T 细胞或降低 MHC-I 的表面表达来逆转。通过遗传或药理学方法用氯喹抑制自噬,与双重 ICB 治疗(抗 PD1 和抗 CTLA4 抗体)协同作用,并导致增强的抗肿瘤免疫反应。我们的研究结果表明,增强的自噬或溶酶体功能通过选择性靶向 MHC-I 分子进行降解在免疫逃逸中起作用,并为自噬抑制和双重 ICB 治疗联合作为治疗 PDAC 的策略提供了依据。