Ramirez Daniel, Kohar Vivek, Lu Mingyang
College of Health Solutions, Arizona State University, Tempe, AZ, United States.
The Jackson Laboratory for Mammalian Genetics, Bar Harbor, ME, United States.
Front Mol Biosci. 2020 Apr 23;7:54. doi: 10.3389/fmolb.2020.00054. eCollection 2020.
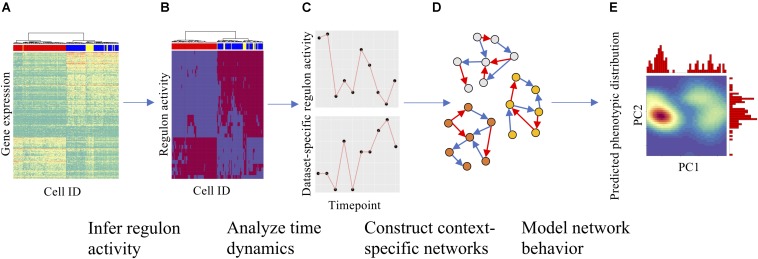
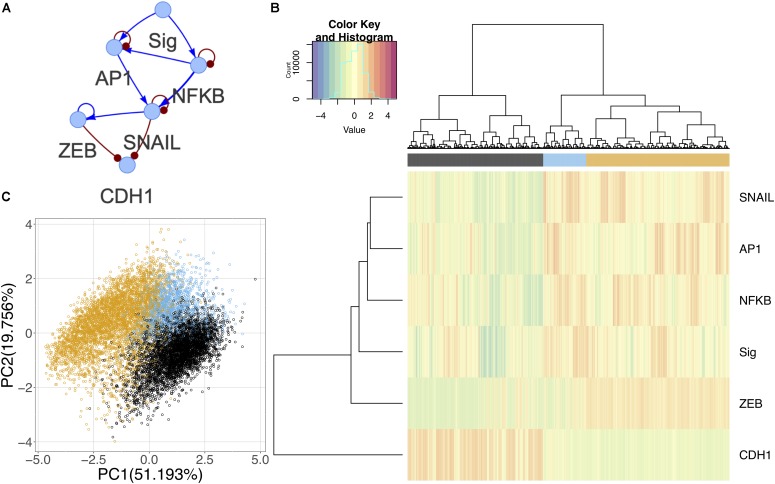
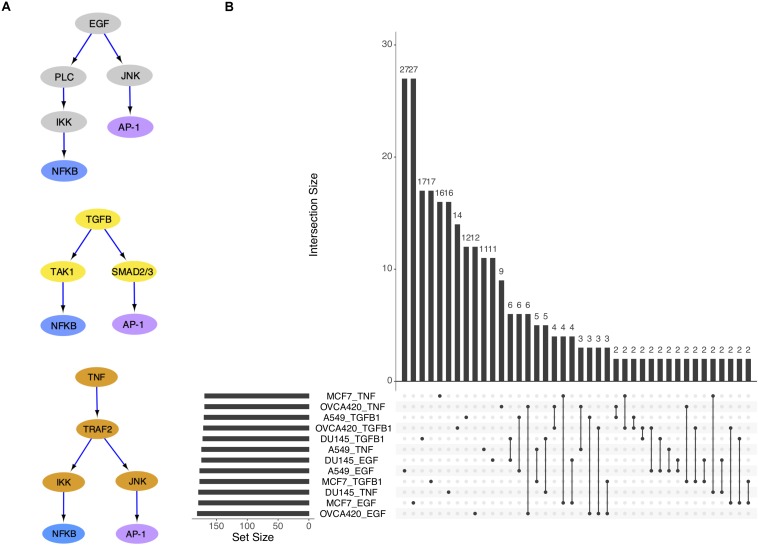
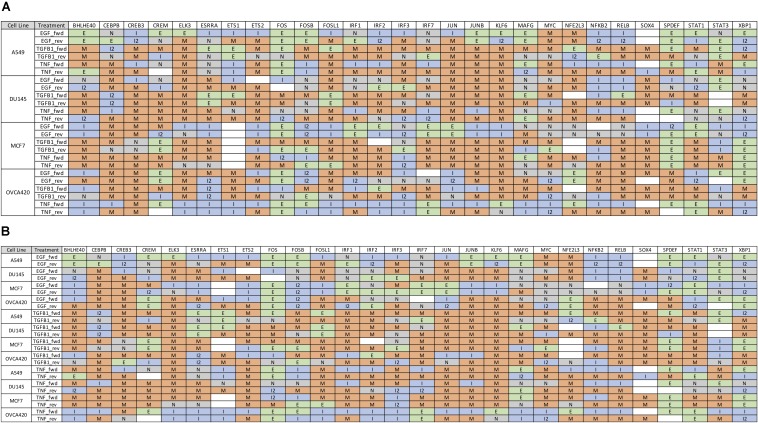
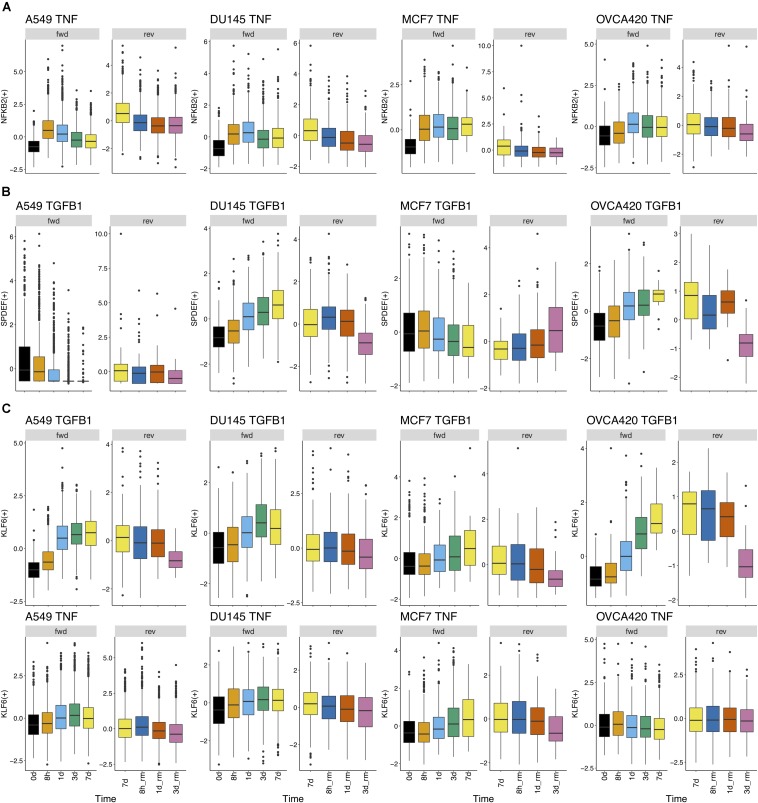
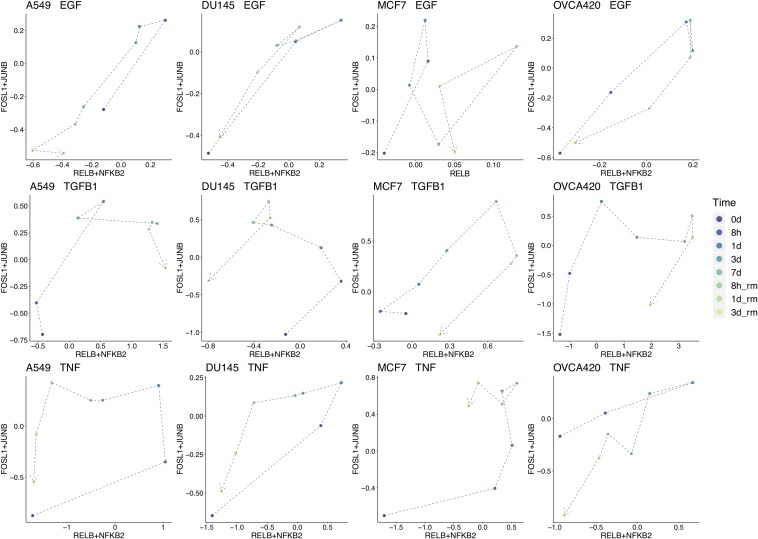
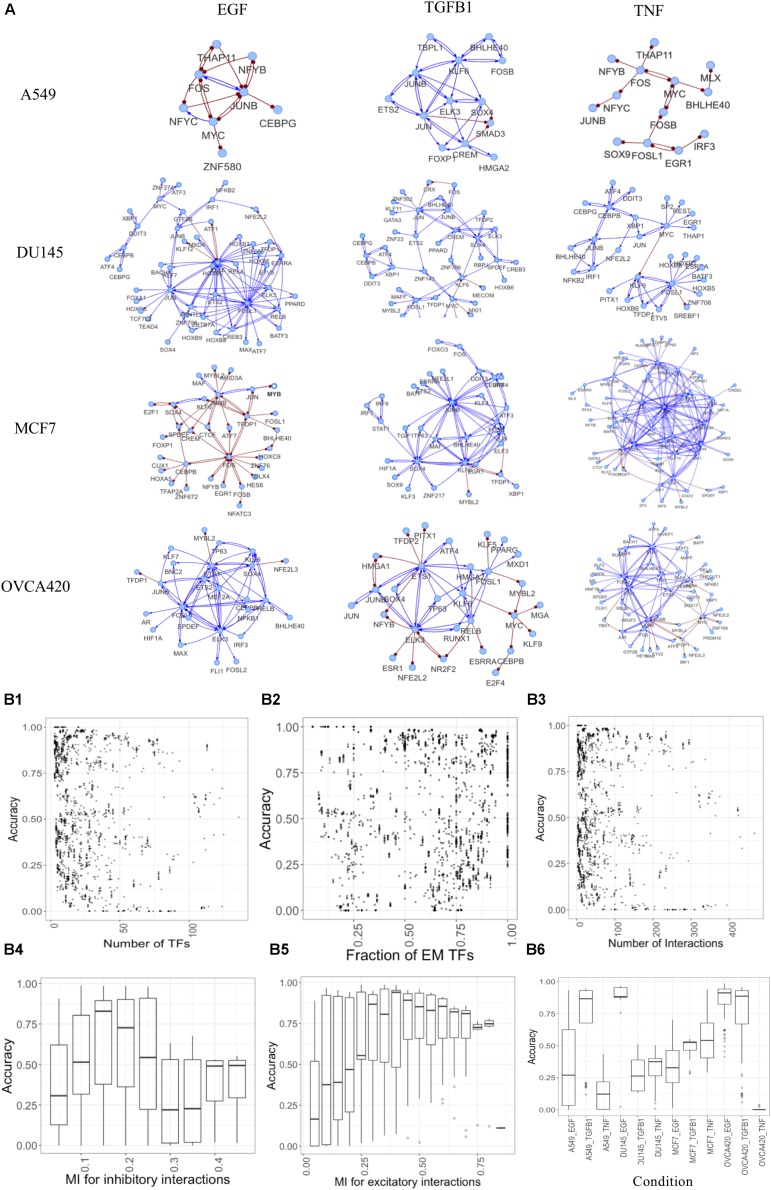
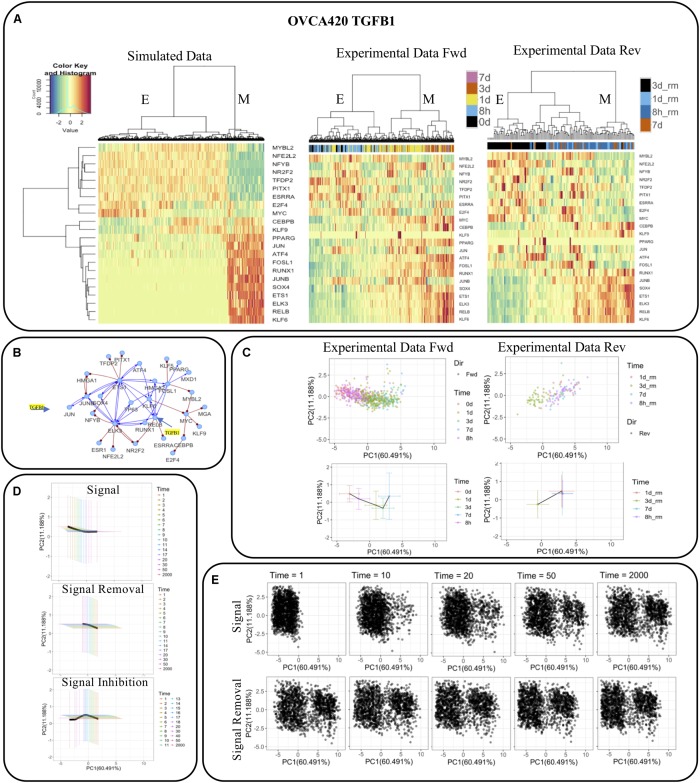
Epithelial-mesenchymal transition (EMT) is well established as playing a crucial role in cancer progression and being a potential therapeutic target. To elucidate the gene regulation that drives the decision making of EMT, many previous studies have been conducted to model EMT gene regulatory circuits (GRCs) using interactions from the literature. While this approach can depict the generic regulatory interactions, it falls short of capturing context-specific features. Here, we explore the effectiveness of a combined bioinformatics and mathematical modeling approach to construct context-specific EMT GRCs directly from transcriptomics data. Using time-series single cell RNA-sequencing data from four different cancer cell lines treated with three EMT-inducing signals, we identify context-specific activity dynamics of common EMT transcription factors. In particular, we observe distinct paths during the forward and backward transitions, as is evident from the dynamics of major regulators such as NF-KB (e.g., NFKB2 and RELB) and AP-1 (e.g., FOSL1 and JUNB). For each experimental condition, we systematically sample a large set of network models and identify the optimal GRC capturing context-specific EMT states using a mathematical modeling method named ndom rcuit rturbation (RACIPE). The results demonstrate that the approach can build high quality GRCs in certain cases, but not others and, meanwhile, elucidate the role of common bioinformatics parameters and properties of network structures in determining the quality of GRCs. We expect the integration of top-down bioinformatics and bottom-up systems biology modeling to be a powerful and generally applicable approach to elucidate gene regulatory mechanisms of cellular state transitions.
上皮-间质转化(EMT)在癌症进展中起着关键作用,并成为一个潜在的治疗靶点,这一点已得到充分证实。为了阐明驱动EMT决策的基因调控,以前进行了许多研究,利用文献中的相互作用来构建EMT基因调控回路(GRC)模型。虽然这种方法可以描绘一般的调控相互作用,但它无法捕捉特定背景下的特征。在这里,我们探索一种结合生物信息学和数学建模的方法直接从转录组学数据构建特定背景下的EMT GRC的有效性。利用来自四种不同癌细胞系并用三种EMT诱导信号处理的时间序列单细胞RNA测序数据,我们确定了常见EMT转录因子的特定背景下的活性动态。特别是,我们观察到正向和反向转变过程中的不同路径,主要调节因子如NF-κB(如NFKB2和RELB)和AP-1(如FOSL1和JUNB)的动态变化就很明显。对于每种实验条件,我们系统地采样大量网络模型,并使用一种名为随机电路扰动(RACIPE)的数学建模方法确定捕捉特定背景下EMT状态的最佳GRC。结果表明,该方法在某些情况下可以构建高质量的GRC,但在其他情况下则不行,同时还阐明了常见生物信息学参数和网络结构属性在确定GRC质量方面的作用。我们期望自上而下的生物信息学和自下而上的系统生物学建模的整合将成为一种强大且普遍适用的方法来阐明细胞状态转变的基因调控机制。