School of Life Sciences, Arizona State University.
Genome Biol Evol. 2020 Jun 1;12(6):871-877. doi: 10.1093/gbe/evaa093.
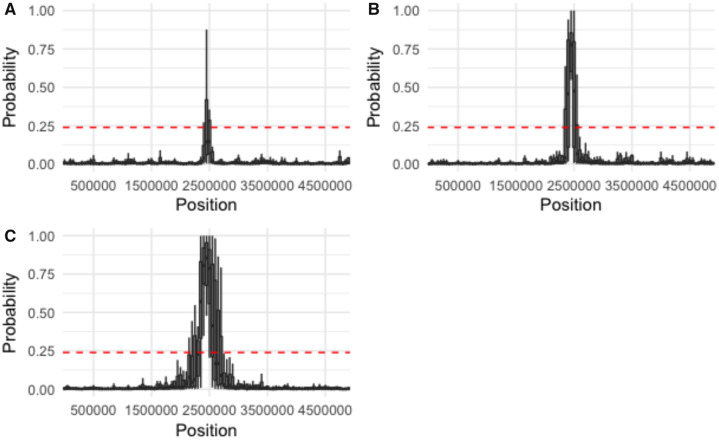
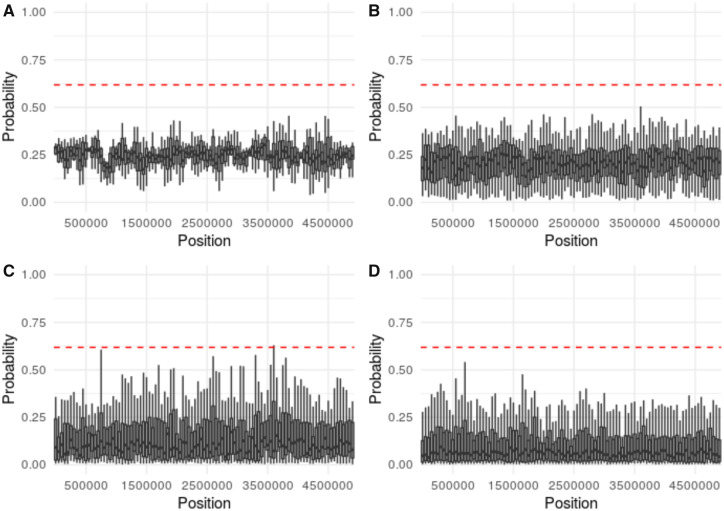
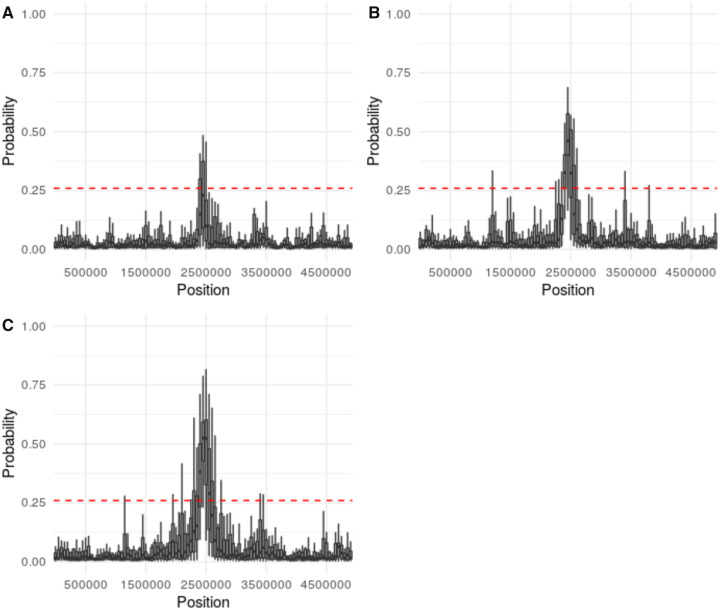
First inspired by the seminal work of Lewontin and Krakauer (1973. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics 74(1):175-195.) and Maynard Smith and Haigh (1974. The hitch-hiking effect of a favourable gene. Genet Res. 23(1):23-35.), genomic scans for positive selection remain a widely utilized tool in modern population genomic analysis. Yet, the relative frequency and genomic impact of selective sweeps have remained a contentious point in the field for decades, largely owing to an inability to accurately identify their presence and quantify their effects-with current methodologies generally being characterized by low true-positive rates and/or high false-positive rates under many realistic demographic models. Most of these approaches are based on Wright-Fisher assumptions and the Kingman coalescent and generally rely on detecting outlier regions which do not conform to these neutral expectations. However, previous theoretical results have demonstrated that selective sweeps are well characterized by an alternative class of model known as the multiple-merger coalescent. Taken together, this suggests the possibility of not simply identifying regions which reject the Kingman, but rather explicitly testing the relative fit of a genomic window to the multiple-merger coalescent. We describe the advantages of such an approach, which owe to the branching structure differentiating selective and neutral models, and demonstrate improved power under certain demographic scenarios relative to a commonly used approach. However, regions of the demographic parameter space continue to exist in which neither this approach nor existing methodologies have sufficient power to detect selective sweeps.
最初受到 Lewontin 和 Krakauer(1973. 基因频率分布作为多态性选择中性理论的检验。遗传学 74(1):175-195.)和 Maynard Smith 和 Haigh(1974. 有利基因的搭便车效应。遗传研究。23(1):23-35.)开创性工作的启发,基因组扫描正向选择仍然是现代群体基因组分析中广泛使用的工具。然而,选择扫荡的相对频率和基因组影响在该领域仍然是一个有争议的问题,这主要是由于无法准确识别它们的存在并量化它们的影响——目前的方法通常具有低真阳性率和/或在许多现实的人口模型下高假阳性率。这些方法大多基于 Wright-Fisher 假设和 Kingman 合并,并且通常依赖于检测不符合这些中性预期的异常区域。然而,先前的理论结果表明,选择扫荡很好地由称为多重合并合并的替代模型类来描述。综上所述,这表明不仅可以识别拒绝 Kingman 的区域,而且可以明确测试基因组窗口对多重合并合并的相对拟合度。我们描述了这种方法的优势,这归因于区分选择和中性模型的分支结构,并证明在某些人口统计场景下相对于常用方法具有更高的功效。然而,在人口统计参数空间的某些区域中,这种方法和现有的方法都没有足够的能力来检测选择扫荡。