Department of Plant Sciences, University of California, Davis, One Shields Avenue, Davis, CA 95616, USA.
Bioinformatics Core Facility, Genome Center, University of California, One Shields Avenue, Davis, CA 95616, USA.
Gigascience. 2020 May 1;9(5). doi: 10.1093/gigascience/giaa050.
The release of the first reference genome of walnut (Juglans regia L.) enabled many achievements in the characterization of walnut genetic and functional variation. However, it is highly fragmented, preventing the integration of genetic, transcriptomic, and proteomic information to fully elucidate walnut biological processes.
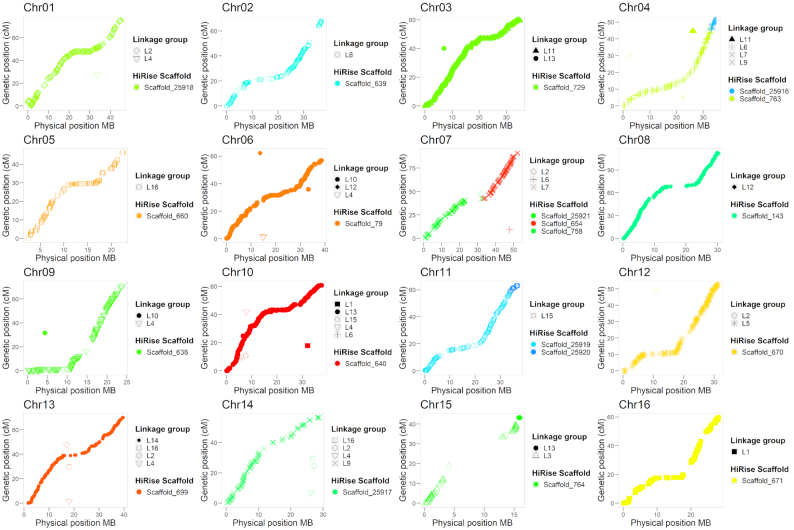
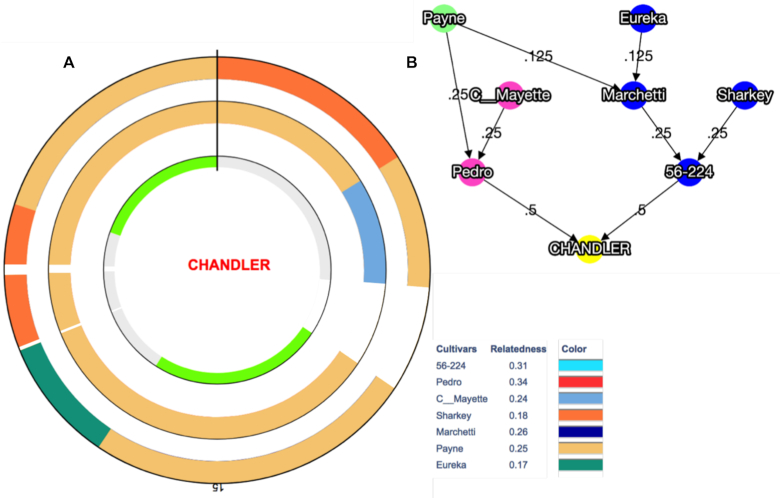
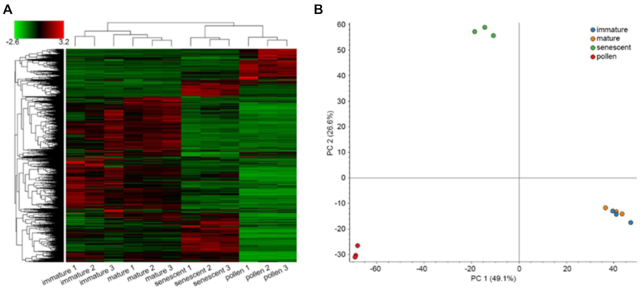
Here, we report the new chromosome-scale assembly of the walnut reference genome (Chandler v2.0) obtained by combining Oxford Nanopore long-read sequencing with chromosome conformation capture (Hi-C) technology. Relative to the previous reference genome, the new assembly features an 84.4-fold increase in N50 size, with the 16 chromosomal pseudomolecules assembled and representing 95% of its total length. Using full-length transcripts from single-molecule real-time sequencing, we predicted 37,554 gene models, with a mean gene length higher than the previous gene annotations. Most of the new protein-coding genes (90%) present both start and stop codons, which represents a significant improvement compared with Chandler v1.0 (only 48%). We then tested the potential impact of the new chromosome-level genome on different areas of walnut research. By studying the proteome changes occurring during male flower development, we observed that the virtual proteome obtained from Chandler v2.0 presents fewer artifacts than the previous reference genome, enabling the identification of a new potential pollen allergen in walnut. Also, the new chromosome-scale genome facilitates in-depth studies of intraspecies genetic diversity by revealing previously undetected autozygous regions in Chandler, likely resulting from inbreeding, and 195 genomic regions highly differentiated between Western and Eastern walnut cultivars.
Overall, Chandler v2.0 will serve as a valuable resource to better understand and explore walnut biology.
核桃(Juglans regia L.)首个参考基因组的发布,使人们在核桃遗传和功能变异的表征方面取得了许多成就。然而,该基因组高度碎片化,阻碍了遗传、转录组和蛋白质组信息的整合,从而无法充分阐明核桃的生物学过程。
本研究通过结合牛津纳米孔长读测序和染色体构象捕获(Hi-C)技术,报道了核桃参考基因组(Chandler v2.0)的新染色体水平组装。与之前的参考基因组相比,新组装的 N50 大小增加了 84.4 倍,16 条染色体假染色体组装并代表其总长度的 95%。利用单分子实时测序的全长转录本,我们预测了 37554 个基因模型,平均基因长度高于之前的基因注释。新的蛋白质编码基因中,90%的基因都有起始和终止密码子,与 Chandler v1.0 相比(仅有 48%)有显著提高。然后,我们测试了新的染色体水平基因组在核桃研究不同领域的潜在影响。通过研究雄性花发育过程中的蛋白质组变化,我们观察到,Chandler v2.0 获得的虚拟蛋白质组比之前的参考基因组产生的伪迹更少,从而能够鉴定出核桃中一种新的潜在花粉过敏原。此外,新的染色体水平基因组通过揭示 Chandler 中以前未检测到的自交区域(可能是由于近亲繁殖),以及 195 个在西方和东方核桃品种之间高度分化的基因组区域,促进了种内遗传多样性的深入研究。
总体而言,Chandler v2.0 将成为更好地理解和探索核桃生物学的宝贵资源。