Department of Cell and Molecular Biology, Tulane University, New Orleans, LA, 70118, USA.
Department of Bioengineering, Rice University, Houston, TX, 77030, USA.
Nat Commun. 2020 May 21;11(1):2549. doi: 10.1038/s41467-020-16312-7.
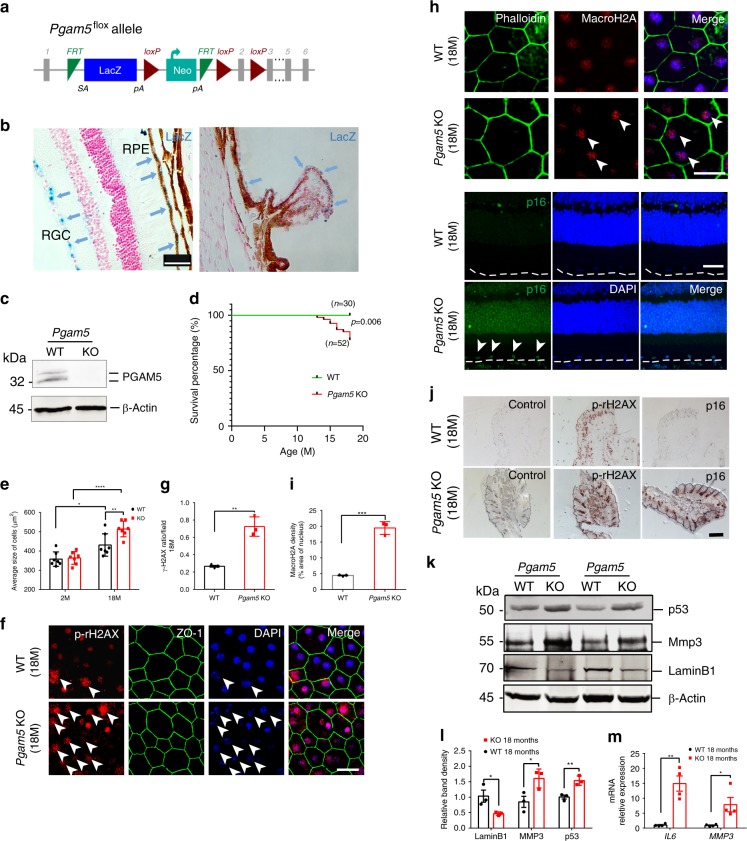
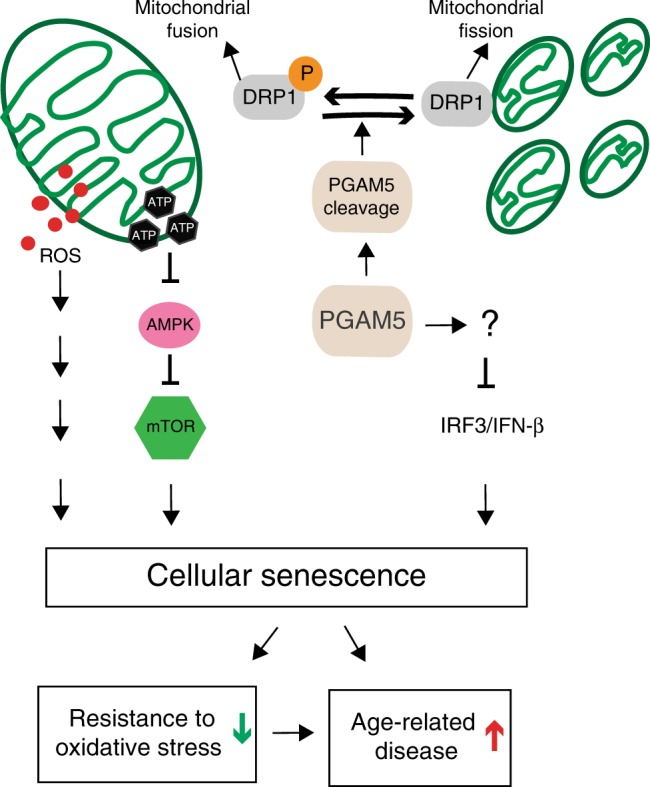
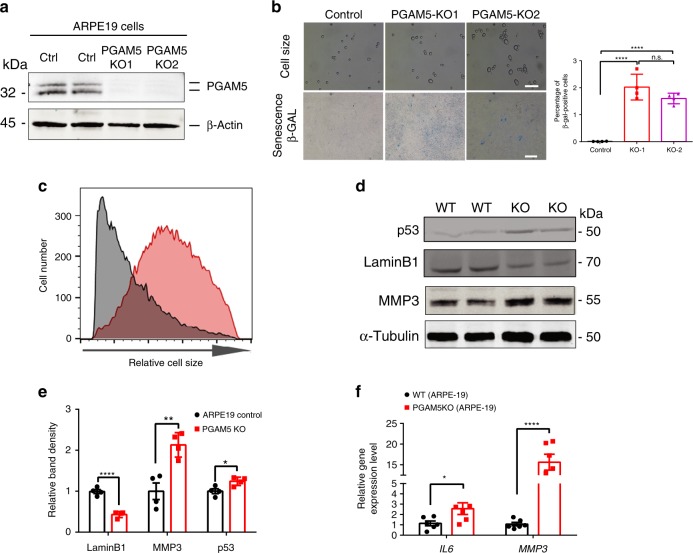
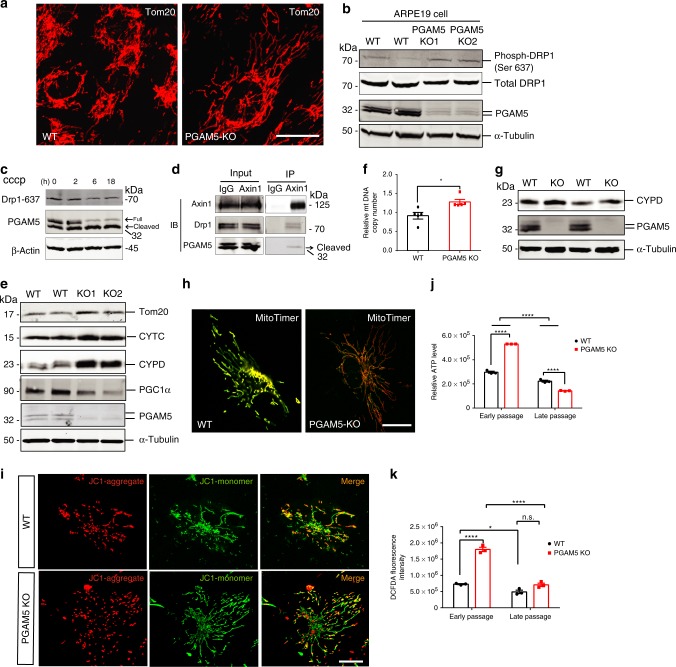
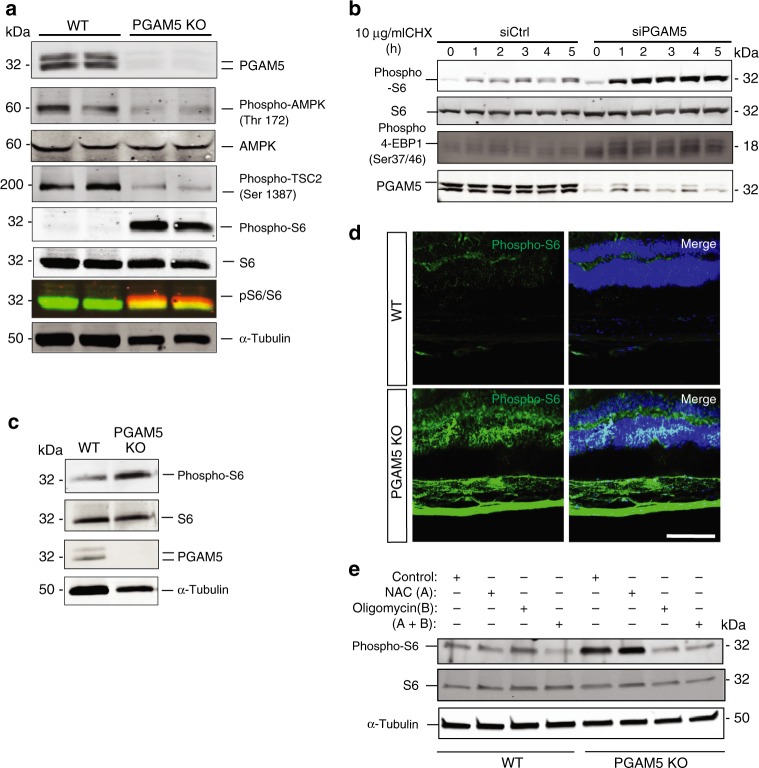
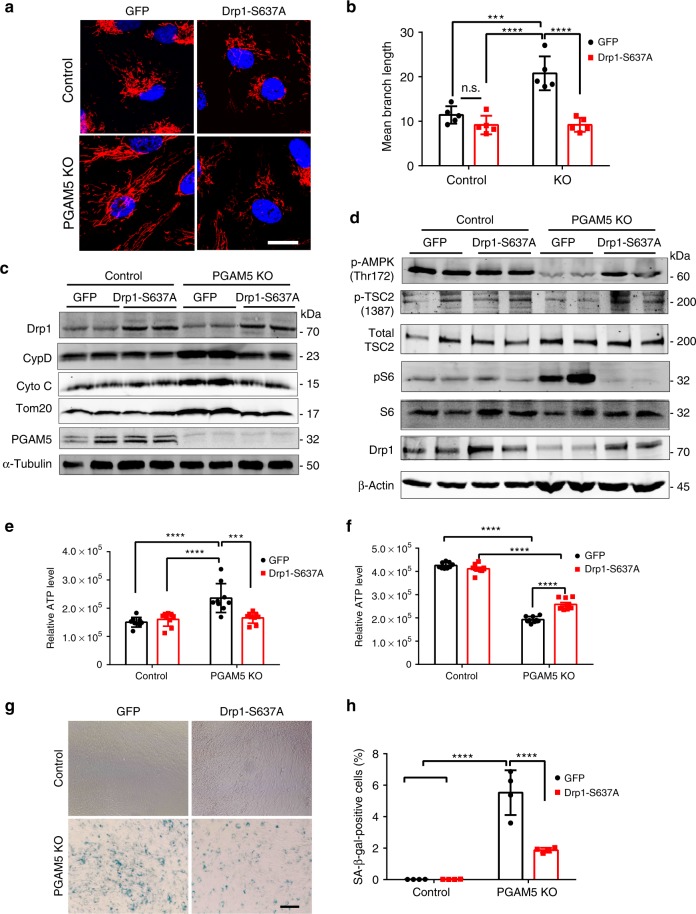
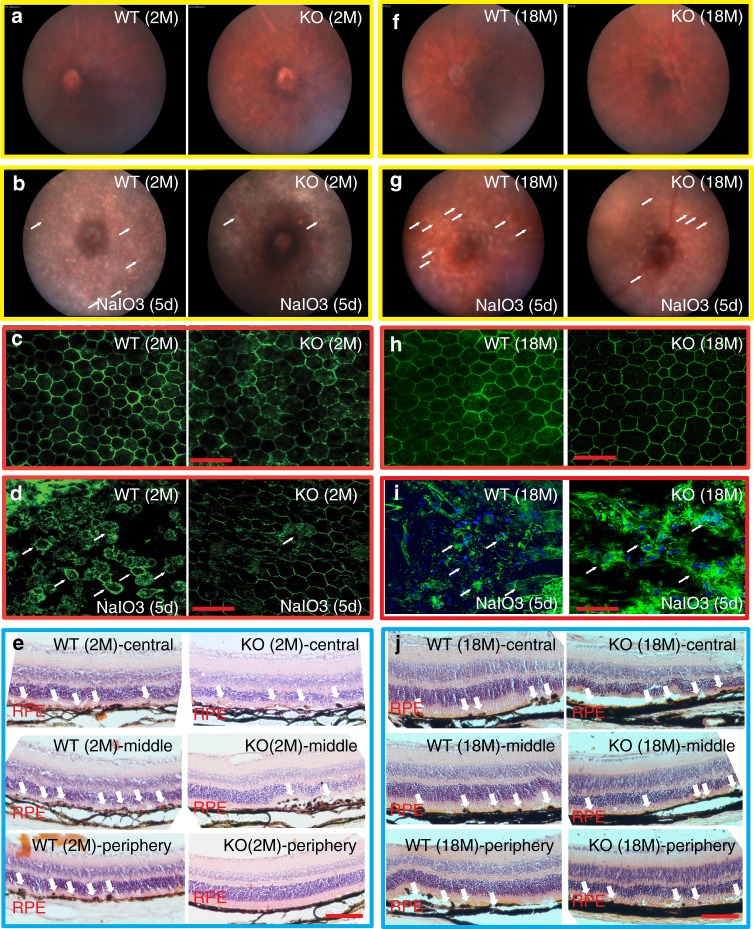
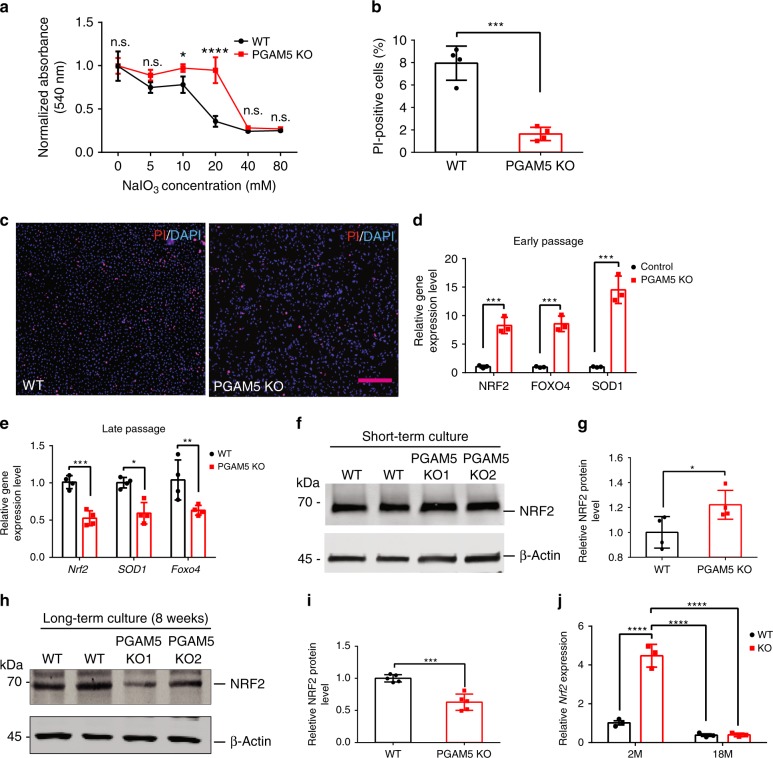
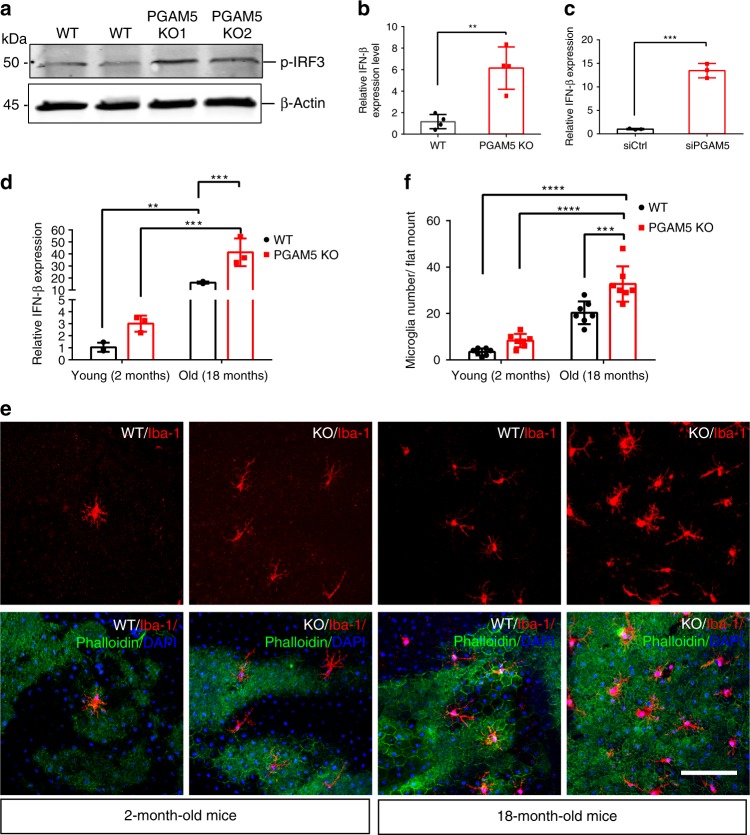
Mitochondria undergo dynamic fusion/fission, biogenesis and mitophagy in response to stimuli or stresses. Disruption of mitochondrial homeostasis could lead to cell senescence, although the underlying mechanism remains unclear. We show that deletion of mitochondrial phosphatase PGAM5 leads to accelerated retinal pigment epithelial (RPE) senescence in vitro and in vivo. Mechanistically, PGAM5 is required for mitochondrial fission through dephosphorylating DRP1. PGAM5 deletion leads to increased mitochondrial fusion and decreased mitochondrial turnover. As results, cellular ATP and reactive oxygen species (ROS) levels are elevated, mTOR and IRF/IFN-β signaling pathways are enhanced, leading to cellular senescence. Overexpression of Drp1 K38A or S637A mutant phenocopies or rescues mTOR activation and senescence in PGAM5 cells, respectively. Young but not aging Pgam5 mice are resistant to sodium iodate-induced RPE cell death. Our studies establish a link between defective mitochondrial fission, cellular senescence and age-dependent oxidative stress response, which have implications in age-related diseases.
线粒体在受到刺激或压力时会发生动态融合/裂变、生物发生和噬线粒体。线粒体动态平衡的破坏可能导致细胞衰老,尽管其潜在机制尚不清楚。我们发现,线粒体磷酸酶 PGAM5 的缺失会导致体外和体内视网膜色素上皮 (RPE) 衰老加速。在机制上,PGAM5 通过去磷酸化 DRP1 来促进线粒体裂变。PGAM5 的缺失会导致线粒体融合增加和线粒体周转率降低。结果,细胞内的 ATP 和活性氧 (ROS) 水平升高,mTOR 和 IRF/IFN-β 信号通路增强,导致细胞衰老。DRp1 K38A 或 S637A 突变体的过表达分别模拟或挽救了 PGAM5 细胞中 mTOR 的激活和衰老。年轻但不老的 Pgam5 小鼠对碘酸钠诱导的 RPE 细胞死亡具有抗性。我们的研究建立了缺陷线粒体裂变、细胞衰老和与年龄相关的氧化应激反应之间的联系,这与与年龄相关的疾病有关。