Instituto Federal de Educação, Ciência e Tecnologia de São Paulo, Catanduva, SP, Brasil.
Departamento de Química, Universidade Federal da Paraíba, Cidade Universitária, João Pessoa, PB, Brasil.
J Biomol Struct Dyn. 2021 Jul;39(11):3924-3933. doi: 10.1080/07391102.2020.1772885. Epub 2020 Jun 2.
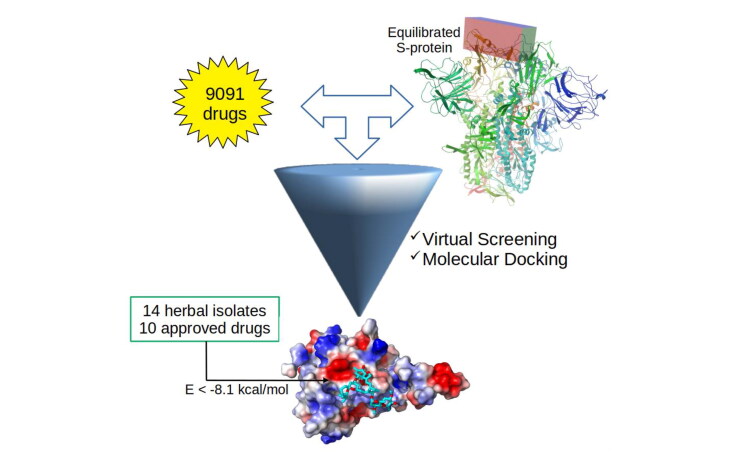
Herein, molecular modeling techniques were used with the main goal to obtain candidates from a drug database as potential targets to be used against SARS-CoV-2. This novel coronavirus, responsible by the COVID-19 outbreak since the end of 2019, became a challenge since there is not vaccine for this disease. The first step in this investigation was to solvate the isolated S-protein in water for molecular dynamics (MD) simulation, being observed a transition from "up" to "down" conformation of receptor-binding domain (RBD) of the S-protein with angle of 54.3 and 43.0 degrees, respectively. The RBD region was more exposed to the solvent and to the possible drugs due to its enhanced surface area. From the equilibrated MD structure, virtual screening by docking calculations were performed using a library contained 9091 FDA approved drugs. Among them, 24 best-scored ligands (14 traditional herbal isolate and 10 approved drugs) with the binding energy below -8.1 kcal/mol were selected as potential candidates to inhibit the SARS-CoV-2 S-protein, preventing the human cell infection and their replication. For instance, the ivermectin drug (present in our list of promise candidates) was recently used successful to control viral replication MD simulations were performed for the three best ligands@S-protein complexes and the binding energies were calculated using the MM/PBSA approach. Overall, it is highlighted an important strategy, some key residues, and chemical groups which may be considered on clinical trials for COVID-19 outbreak. [Formula: see text]Communicated by Ramaswamy H. Sarma.
在此,我们使用分子建模技术,主要目的是从药物数据库中获得候选物,作为针对 SARS-CoV-2 的潜在靶点。这种新型冠状病毒是导致 2019 年底 COVID-19 爆发的罪魁祸首,由于目前尚无针对该疾病的疫苗,因此成为了一个挑战。这项研究的第一步是将分离的 S 蛋白在水中进行分子动力学(MD)模拟,观察到 S 蛋白受体结合域(RBD)从“向上”构象到“向下”构象的转变,角度分别为 54.3 和 43.0 度。由于 RBD 区域的表面积增加,因此更容易暴露于溶剂和可能的药物中。从平衡的 MD 结构中,使用包含 9091 种已批准用于 FDA 的药物的库进行虚拟筛选对接计算。其中,选择了 24 种评分最高的配体(14 种传统草药分离物和 10 种已批准的药物),它们的结合能低于-8.1 kcal/mol,作为潜在的候选物,以抑制 SARS-CoV-2 S 蛋白,防止人类细胞感染及其复制。例如,伊维菌素药物(在我们有希望的候选药物名单中)最近成功用于控制病毒复制。对三种最佳配体@S 蛋白复合物进行 MD 模拟,并使用 MM/PBSA 方法计算结合能。总体而言,强调了一种重要的策略、一些关键残基和化学基团,这些可能会在 COVID-19 爆发的临床试验中得到考虑。 [公式:见正文]由 Ramaswamy H. Sarma 交流。