Division of Vaccine Discovery, La Jolla Institute for Immunology, La Jolla, California, United States of America.
Infection and Immunity Program, Biomedicine Discovery Institute, Monash University, Clayton, VIC, Australia.
PLoS Comput Biol. 2020 May 26;16(5):e1007757. doi: 10.1371/journal.pcbi.1007757. eCollection 2020 May.
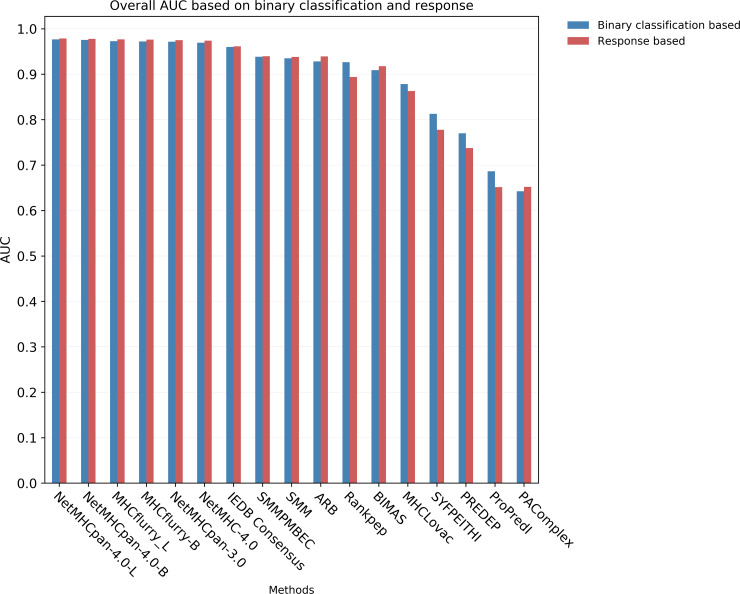
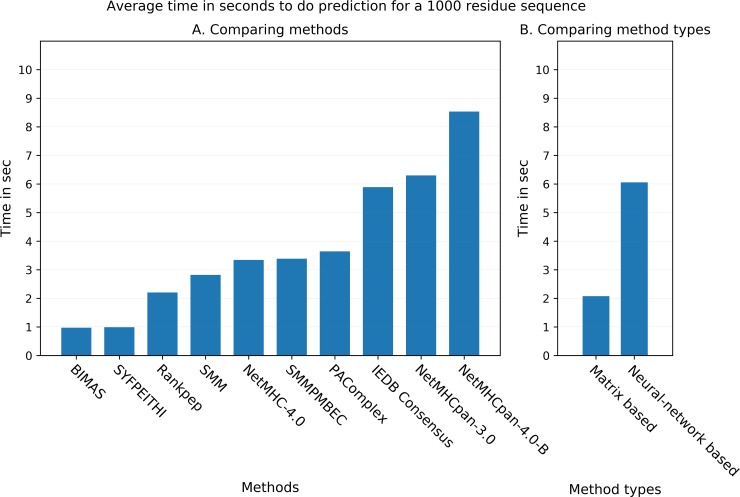
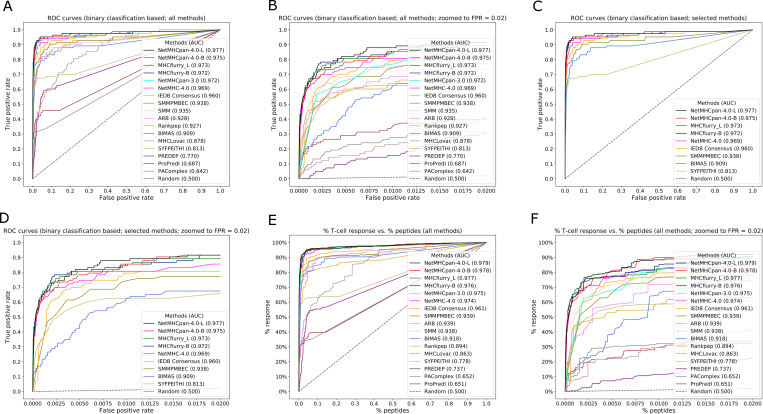
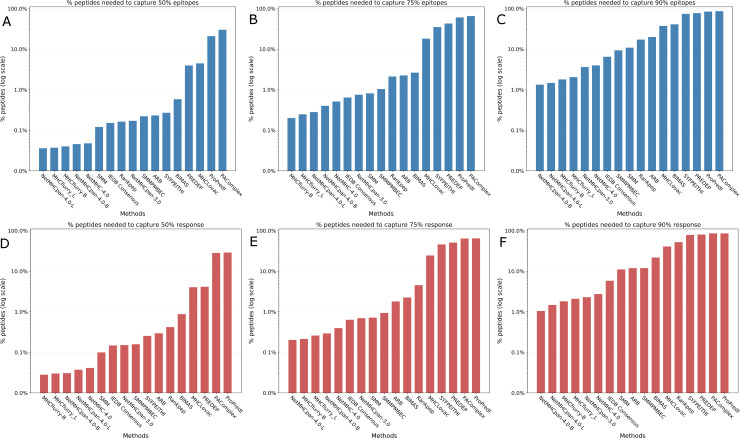
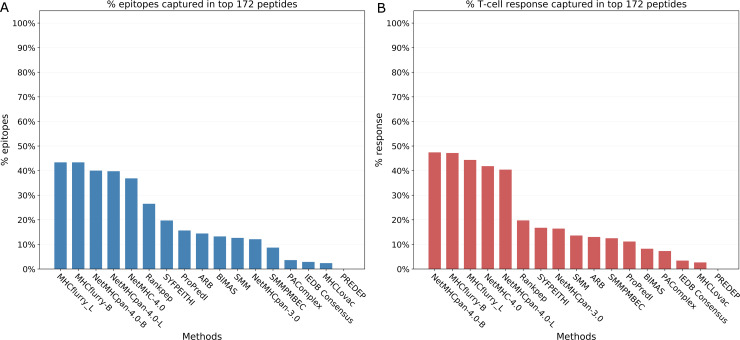
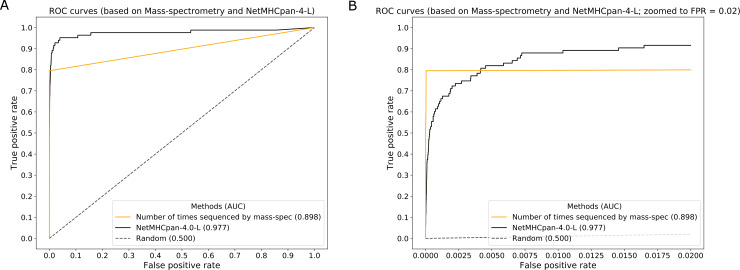
T cell epitope candidates are commonly identified using computational prediction tools in order to enable applications such as vaccine design, cancer neoantigen identification, development of diagnostics and removal of unwanted immune responses against protein therapeutics. Most T cell epitope prediction tools are based on machine learning algorithms trained on MHC binding or naturally processed MHC ligand elution data. The ability of currently available tools to predict T cell epitopes has not been comprehensively evaluated. In this study, we used a recently published dataset that systematically defined T cell epitopes recognized in vaccinia virus (VACV) infected C57BL/6 mice (expressing H-2Db and H-2Kb), considering both peptides predicted to bind MHC or experimentally eluted from infected cells, making this the most comprehensive dataset of T cell epitopes mapped in a complex pathogen. We evaluated the performance of all currently publicly available computational T cell epitope prediction tools to identify these major epitopes from all peptides encoded in the VACV proteome. We found that all methods were able to improve epitope identification above random, with the best performance achieved by neural network-based predictions trained on both MHC binding and MHC ligand elution data (NetMHCPan-4.0 and MHCFlurry). Impressively, these methods were able to capture more than half of the major epitopes in the top N = 277 predictions within the N = 767,788 predictions made for distinct peptides of relevant lengths that can theoretically be encoded in the VACV proteome. These performance metrics provide guidance for immunologists as to which prediction methods to use, and what success rates are possible for epitope predictions when considering a highly controlled system of administered immunizations to inbred mice. In addition, this benchmark was implemented in an open and easy to reproduce format, providing developers with a framework for future comparisons against new tools.
T 细胞表位候选物通常使用计算预测工具来识别,以便能够应用于疫苗设计、癌症新抗原识别、诊断开发和去除针对蛋白质治疗的不必要免疫反应等领域。大多数 T 细胞表位预测工具都是基于针对 MHC 结合或天然加工的 MHC 配体洗脱数据进行训练的机器学习算法。目前可用的工具预测 T 细胞表位的能力尚未得到全面评估。在这项研究中,我们使用了最近发表的数据集,该数据集系统地定义了在表达 H-2Db 和 H-2Kb 的 C57BL/6 小鼠中感染牛痘病毒 (VACV) 时识别的 T 细胞表位,同时考虑了预测与 MHC 结合或从感染细胞中洗脱的肽,这是在复杂病原体中映射 T 细胞表位的最全面数据集。我们评估了所有当前公开可用的计算 T 细胞表位预测工具的性能,以从 VACV 蛋白质组中所有编码的肽中识别这些主要表位。我们发现,所有方法都能够提高表位识别的准确性,超过了随机水平,基于 MHC 结合和 MHC 配体洗脱数据训练的神经网络预测方法(NetMHCPan-4.0 和 MHCFlurry)表现最好。令人印象深刻的是,这些方法能够在理论上可编码在 VACV 蛋白质组中的相关长度的独特肽的 N = 767788 个预测中,在 N = 277 个预测中捕获超过一半的主要表位。这些性能指标为免疫学家提供了指导,说明在考虑对近交系小鼠进行高度控制的免疫接种时,应使用哪种预测方法以及进行表位预测的成功率是多少。此外,该基准以开放且易于重现的格式实现,为开发人员提供了一个与新工具进行未来比较的框架。