Laboratoire d'Optique et Biosciences, Ecole Polytechnique, IP Paris, F-91128 Palaiseau, France.
Department of Biochemistry and Molecular Biology, Gordon Center for Integrative Science, University of Chicago, 929 E57th Street, Chicago, Illinois 60637, United States.
J Chem Theory Comput. 2020 Jul 14;16(7):4655-4668. doi: 10.1021/acs.jctc.0c00111. Epub 2020 Jun 12.
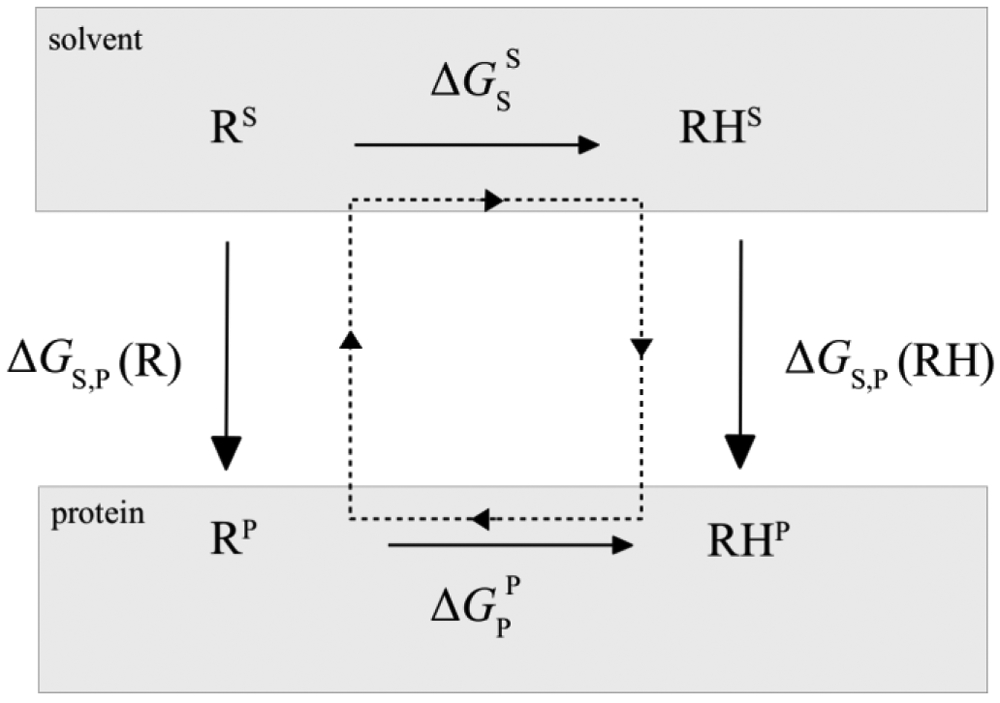
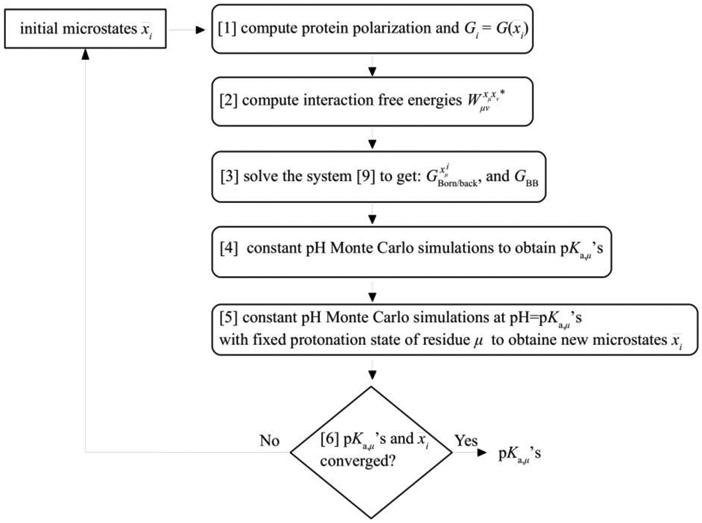
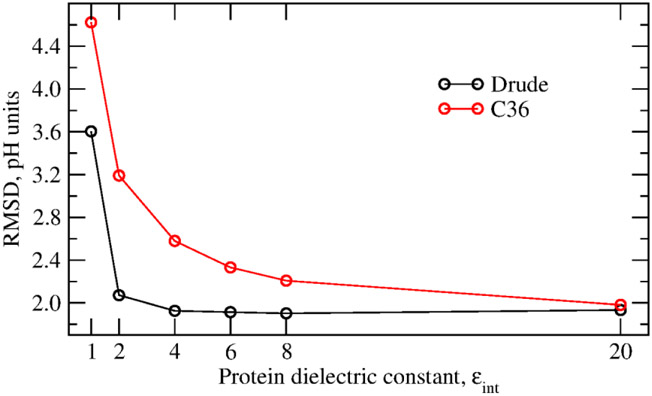
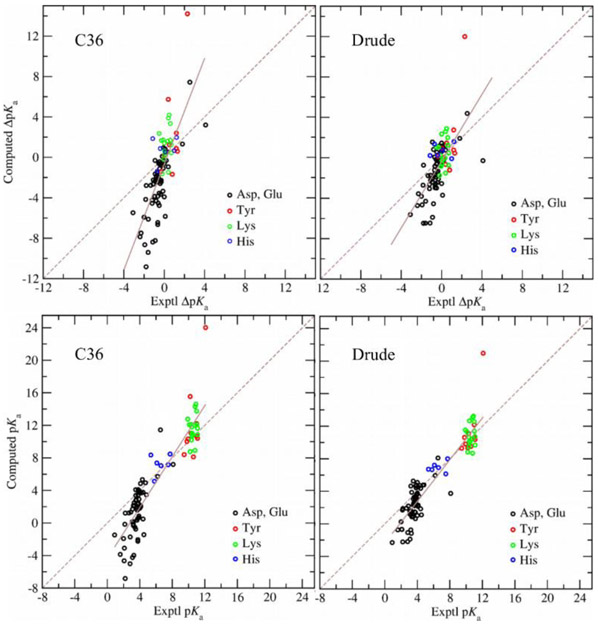
Electronic polarization effects have been suggested to play an important role in proton binding to titratable residues in proteins. In this work, we describe a new computational method for p calculations, using Monte Carlo (MC) simulations to sample protein protonation states with the Drude polarizable force field and Poisson-Boltzmann (PB) continuum electrostatic solvent model. While the most populated protonation states at the selected pH, corresponding to residues that are half-protonated at that pH, are sampled using the exact relative free energies computed with Drude particles optimized in the field of the PB implicit solvation model, we introduce an approximation for the protein polarization of low-populated protonation states to reduce the computational cost. The highly populated protonation states used to compute the polarization and p's are then iteratively improved until convergence. It is shown that for lysozyme, when considering 9 of the 18 titratable residues, the new method converged within two iterations with computed p's differing only by 0.02 pH units from p's estimated with the exact approach. Application of the method to predict p's of 94 titratable side chains in 8 proteins shows the Drude-PB model to produce physically more correct results as compared to the additive CHARMM36 (C36) force field (FF). With a dielectric constant of two assigned to the protein interior the Root Mean Square (RMS) deviation between computed and experimental p's is 2.07 and 3.19 pH units with the Drude and C36 models, respectively, and the RMS deviation using the Drude-PB model is relatively insensitive to the choice of the internal dielectric constant in contrast to the additive C36 model. At the higher internal dielectric constant of 20, p's computed with the additive C36 model converge to the results obtained with the Drude polarizable force field, indicating the need to artificially overestimate electrostatic screening in a nonphysical way with the additive FF. In addition, inclusion of both and orientations of the proton in the neutral state of acidic groups is shown to yield improved agreement with experiment. The present work, which is the first example of the use of a polarizable model for the prediction of p's in proteins, shows that the use of a polarizable model represents a more physically correct model for the treatment of electrostatic contributions to p shifts in proteins.
电子极化效应对质子与蛋白质中可滴定残基的结合起着重要作用。在这项工作中,我们描述了一种新的 p 值计算方法,使用蒙特卡罗(MC)模拟,用 Drude 极化力场和泊松-玻尔兹曼(PB)连续静电溶剂模型来采样蛋白质质子化状态。在选定的 pH 值下,最常出现的质子化状态,对应于该 pH 值下半质子化的残基,是使用在 PB 隐式溶剂模型的场中优化的 Drude 粒子计算的精确相对自由能来采样的,我们引入了一种对低占据质子化状态的蛋白质极化的近似方法,以降低计算成本。用于计算极化和 p 值的高占据质子化状态然后进行迭代改进,直到收敛。结果表明,对于溶菌酶,当考虑 18 个可滴定残基中的 9 个时,新方法在两次迭代内收敛,计算得到的 p 值与使用精确方法估计的 p 值相差仅 0.02 pH 单位。该方法应用于预测 8 种蛋白质中 94 个可滴定侧链的 p 值,表明 Drude-PB 模型比加和 CHARMM36(C36)力场(FF)产生更符合物理实际的结果。在蛋白质内部的介电常数为 2 的情况下,计算得到的 p 值与实验 p 值的均方根(RMS)偏差分别为 2.07 和 3.19 pH 单位,与 Drude 和 C36 模型相比,使用 Drude-PB 模型的 RMS 偏差相对不敏感,与加和 C36 模型不同,后者的内部介电常数的选择。在较高的内部介电常数 20 下,使用加和 C36 模型计算的 p 值收敛到与 Drude 极化力场计算得到的结果,这表明需要以非物理的方式人为地夸大加和 FF 中的静电屏蔽作用。此外,在酸性基团的中性状态下包含质子的 和 两种取向,会提高与实验结果的一致性。本工作是首次在蛋白质中 p 值预测中使用极化模型的实例,表明使用极化模型代表了一种更符合物理实际的处理蛋白质中 p 值变化的静电贡献的模型。