Computational Molecular Evolution Group, Heidelberg Institute for Theoretical Studies, Heidelberg, Germany.
Institute for Theoretical Informatics, Karlsruhe Institute of Technology, Karlsruhe, Germany.
Mol Biol Evol. 2020 Sep 1;37(9):2763-2774. doi: 10.1093/molbev/msaa141.
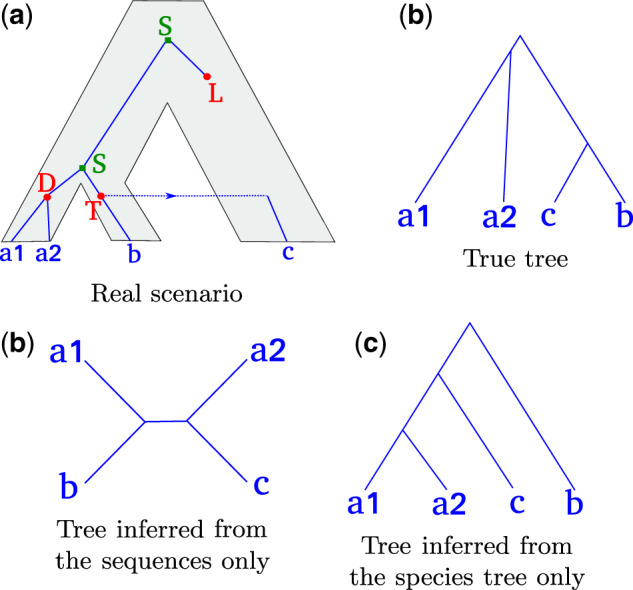
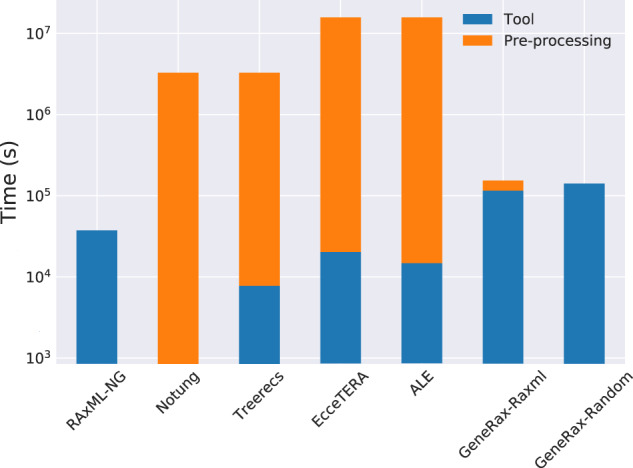
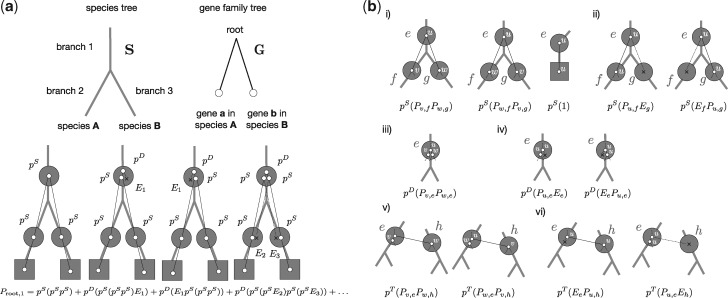
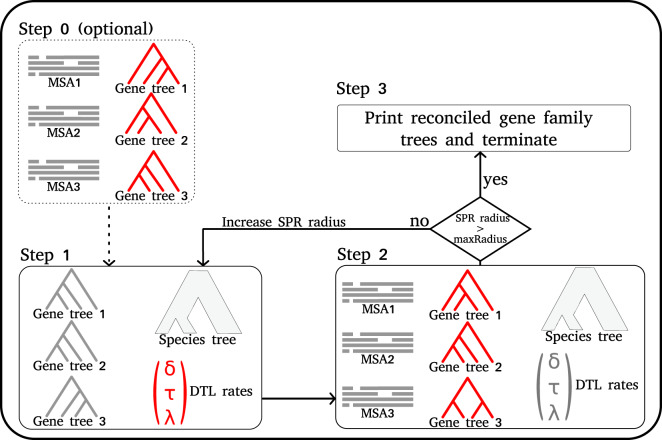
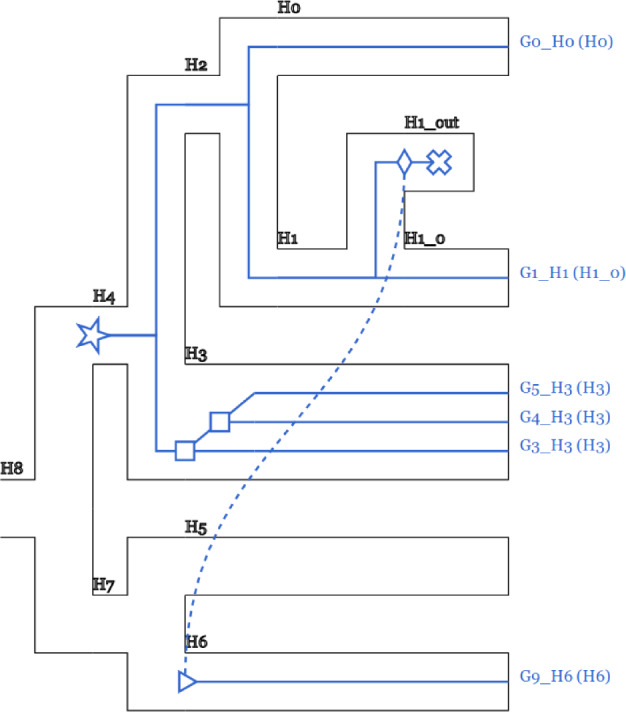
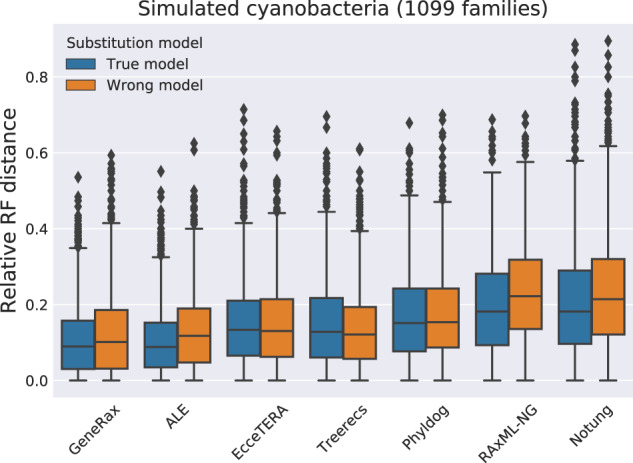
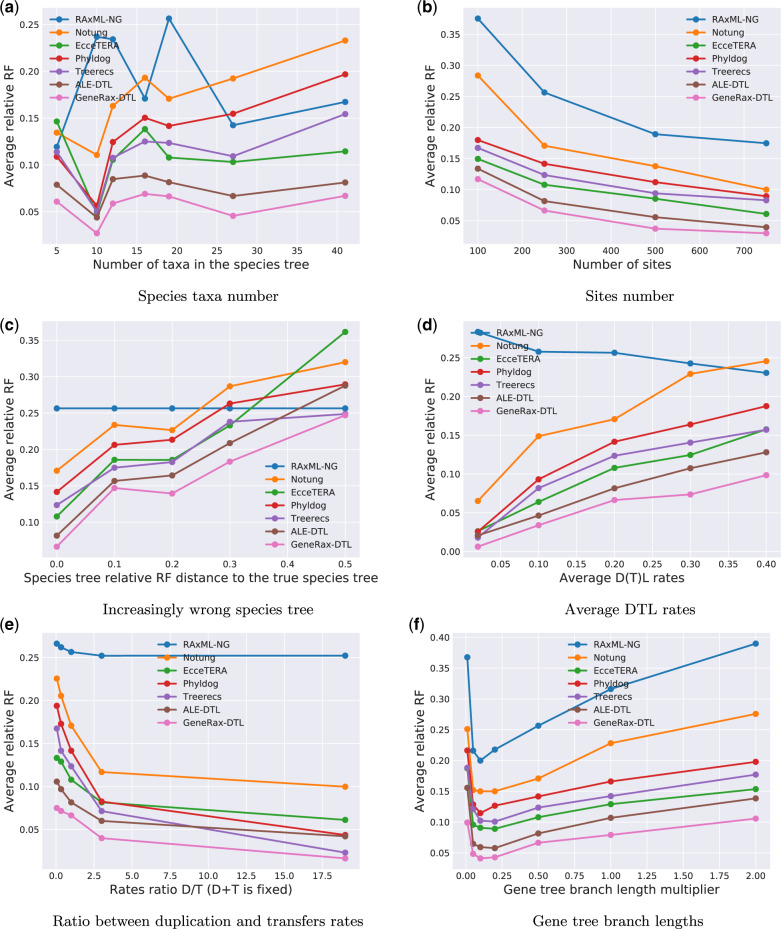
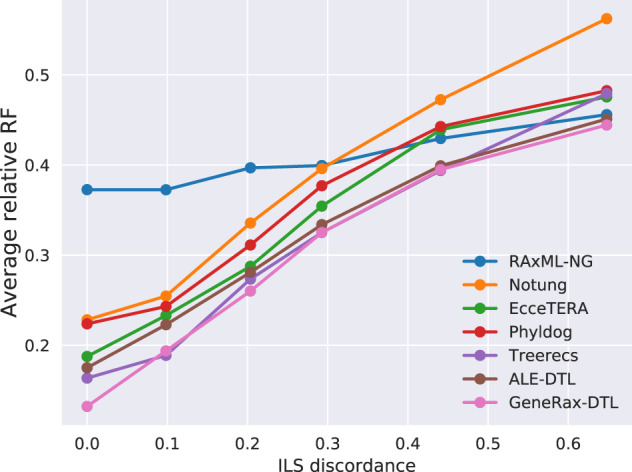
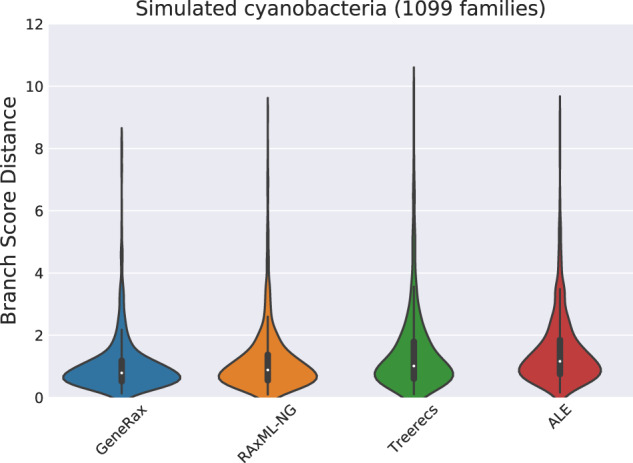
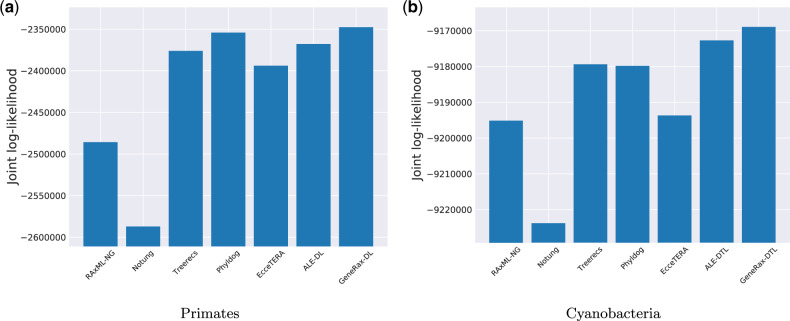
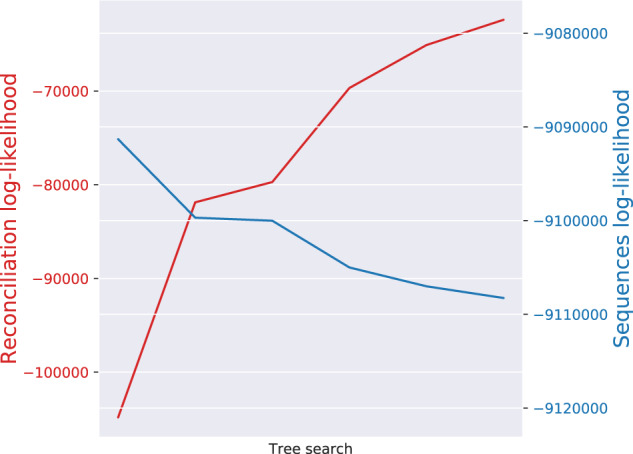
Inferring phylogenetic trees for individual homologous gene families is difficult because alignments are often too short, and thus contain insufficient signal, while substitution models inevitably fail to capture the complexity of the evolutionary processes. To overcome these challenges, species-tree-aware methods also leverage information from a putative species tree. However, only few methods are available that implement a full likelihood framework or account for horizontal gene transfers. Furthermore, these methods often require expensive data preprocessing (e.g., computing bootstrap trees) and rely on approximations and heuristics that limit the degree of tree space exploration. Here, we present GeneRax, the first maximum likelihood species-tree-aware phylogenetic inference software. It simultaneously accounts for substitutions at the sequence level as well as gene level events, such as duplication, transfer, and loss relying on established maximum likelihood optimization algorithms. GeneRax can infer rooted phylogenetic trees for multiple gene families, directly from the per-gene sequence alignments and a rooted, yet undated, species tree. We show that compared with competing tools, on simulated data GeneRax infers trees that are the closest to the true tree in 90% of the simulations in terms of relative Robinson-Foulds distance. On empirical data sets, GeneRax is the fastest among all tested methods when starting from aligned sequences, and it infers trees with the highest likelihood score, based on our model. GeneRax completed tree inferences and reconciliations for 1,099 Cyanobacteria families in 8 min on 512 CPU cores. Thus, its parallelization scheme enables large-scale analyses. GeneRax is available under GNU GPL at https://github.com/BenoitMorel/GeneRax (last accessed June 17, 2020).
推断单个同源基因家族的系统发育树是困难的,因为比对通常太短,因此包含的信号不足,而替代模型不可避免地无法捕捉到进化过程的复杂性。为了克服这些挑战,种系树感知方法还利用了假定种系树的信息。然而,只有少数方法可用,这些方法实现了完整的似然框架或考虑了水平基因转移。此外,这些方法通常需要昂贵的数据预处理(例如,计算引导树),并且依赖于限制树空间探索程度的近似值和启发式方法。在这里,我们介绍了 GeneRax,这是第一个最大似然种系树感知系统发育推断软件。它同时考虑了序列水平和基因水平的替代,例如基于已建立的最大似然优化算法的复制、转移和丢失。GeneRax 可以从每个基因的序列比对和一个有根但未标记的种系树直接推断出多个基因家族的有根系统发育树。我们表明,与竞争工具相比,在模拟数据中,在 90%的模拟中,GeneRax 推断的树在相对罗宾逊-福尔兹距离方面最接近真实树。在实际数据集上,从对齐序列开始时,GeneRax 是所有测试方法中最快的,并且根据我们的模型推断出具有最高似然评分的树。GeneRax 在 512 个 CPU 内核上用 8 分钟完成了 1099 个蓝藻家族的树推断和协调。因此,其并行化方案支持大规模分析。GeneRax 在 https://github.com/BenoitMorel/GeneRax 下根据 GNU GPL 提供(最后访问时间为 2020 年 6 月 17 日)。