Biomedical Sciences Graduate Program, University of California San Diego, La Jolla, CA, 92093-0419, USA.
Department of Biomedical Informatics, University of California San Diego, La Jolla, CA, 92093-0419, USA.
Nat Commun. 2020 Jun 10;11(1):2928. doi: 10.1038/s41467-020-16481-5.
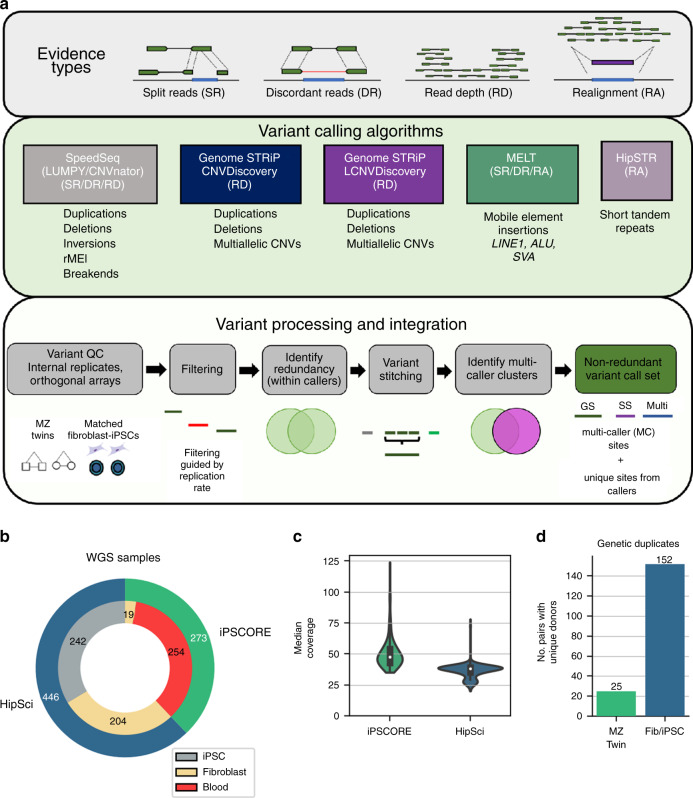
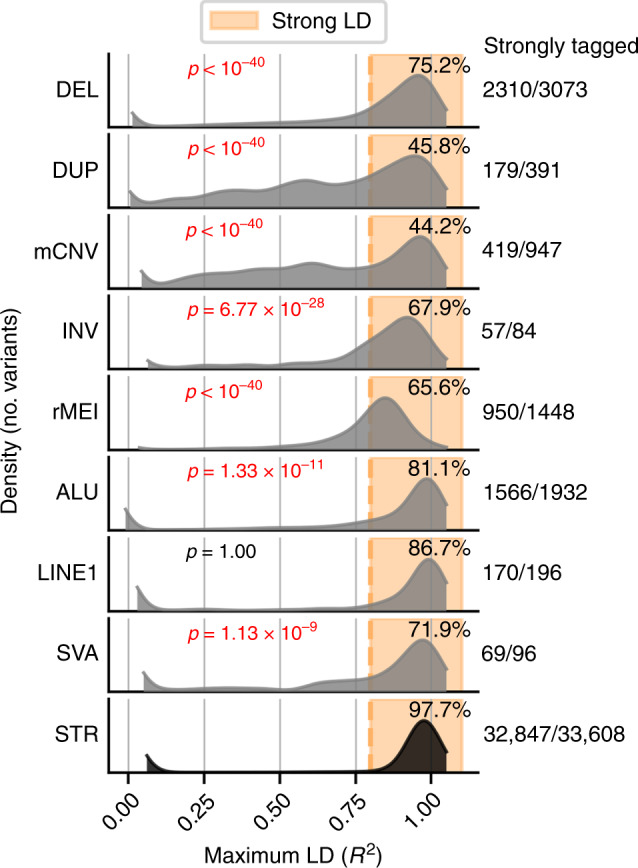
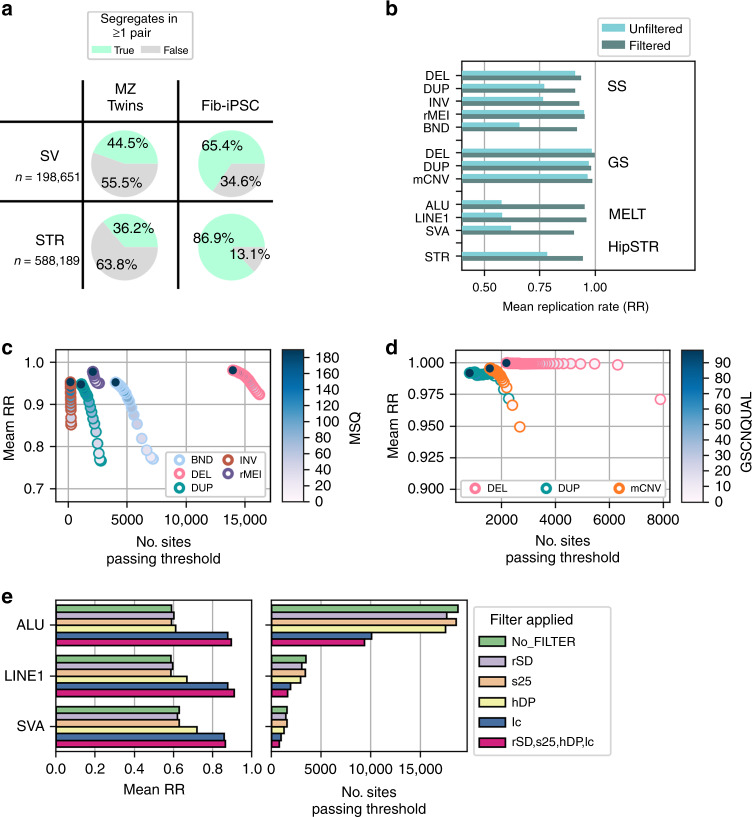
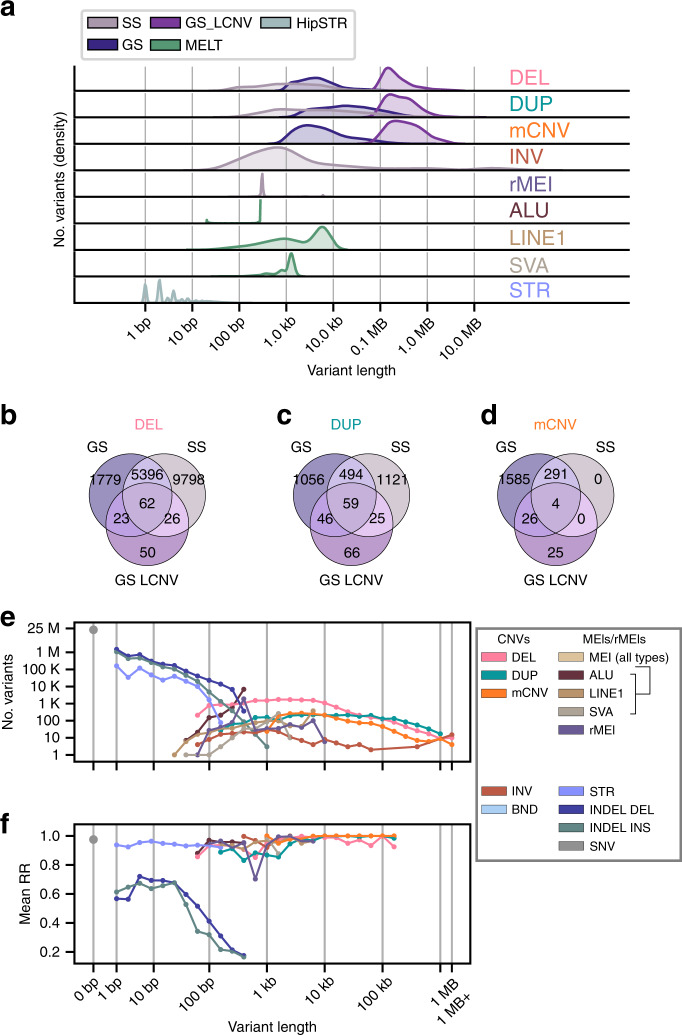
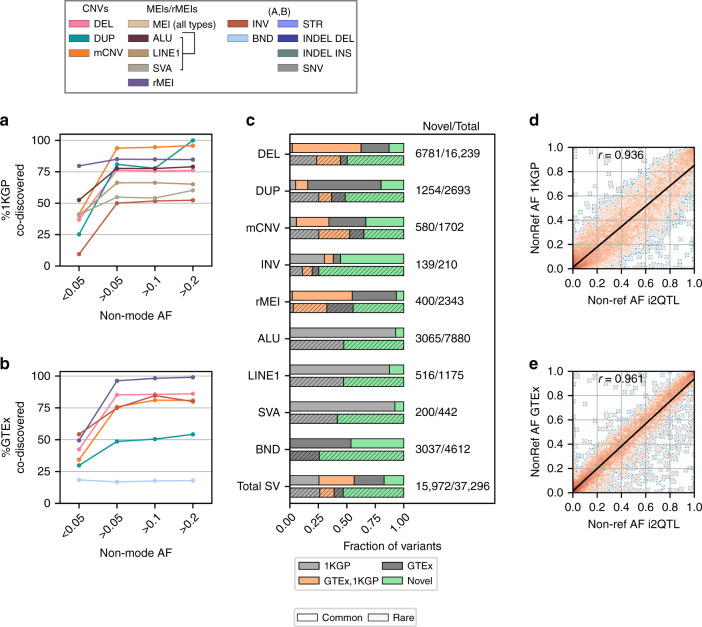
Structural variants (SVs) and short tandem repeats (STRs) are important sources of genetic diversity but are not routinely analyzed in genetic studies because they are difficult to accurately identify and genotype. Because SVs and STRs range in size and type, it is necessary to apply multiple algorithms that incorporate different types of evidence from sequencing data and employ complex filtering strategies to discover a comprehensive set of high-quality and reproducible variants. Here we assemble a set of 719 deep whole genome sequencing (WGS) samples (mean 42×) from 477 distinct individuals which we use to discover and genotype a wide spectrum of SV and STR variants using five algorithms. We use 177 unique pairs of genetic replicates to identify factors that affect variant call reproducibility and develop a systematic filtering strategy to create of one of the most complete and well characterized maps of SVs and STRs to date.
结构变异(SV)和短串联重复(STR)是遗传多样性的重要来源,但由于难以准确识别和基因分型,通常不在遗传研究中进行分析。由于 SV 和 STR 的大小和类型不同,因此需要应用多种算法,这些算法结合了测序数据中不同类型的证据,并采用复杂的过滤策略来发现一套全面的高质量且可重复的变体。在这里,我们组装了一组来自 477 个不同个体的 719 个深度全基因组测序(WGS)样本(平均 42×),我们使用这组样本,通过五种算法来发现和基因分型各种 SV 和 STR 变体。我们使用 177 对独特的遗传重复样本,来确定影响变体调用可重复性的因素,并开发了一种系统的过滤策略,以创建迄今为止最完整和特征描述最好的 SV 和 STR 图谱之一。